
A leucodistrofia metacromática (MLD) é uma doença neurodegenerativa rara e progressiva que afeta o sistema nervoso central. É causada pela deficiência de uma enzima chamada arilsulfatase A (ARSA), que é responsável pela quebra de um tipo de lipídio chamado sulfatide. O acúmulo desse no sistema nervoso central causa desmielinização. A prevalência varia de 1:40.000 a 1:100.000. A doença tem três formas: infantil, juvenil e adulta, sendo que a forma infantil é a mais grave e a forma adulta é a mais leve. Os sintomas variam dependendo da forma da doença, destacam-se as manifestações neurocognitivas e motoras1. A forma infantil é a mais grave e ocorre nos primeiros anos de vida. Os sintomas começam entre os 18 meses e os 3 anos de idade e incluem atraso no desenvolvimento, problemas de coordenação, convulsões, paralisia e demência.
Os achados de imagem mais característicos da doença são identificados em estudos de ressonância nuclear magnética de crânio (RNM) evidenciando hiperintensidades da substância branca periventricular. Na tomografia de crânio observam-se hiperdensidades da substância branca, principalmente nas regiões frontal e parietal. O diagnóstico definitivo é bioquímico e genético. No sangue e urina dosa-se nível anormalmente baixo de arilsulfatase A2,3.
O objetivo deste artigo é relatar o raro caso pediátrico de leucodistrofia metacromática na forma infantil.
RELATO DE CASO
O estudo foi aprovado pelo comitê de Ética e Pesquisa do Hospital Pequeno Príncipe (CAEE: 64291122.0.0000.0097). Os familiares da paciente assinaram o Termo de Consentimento Livre e Esclarecido para utilização das imagens e informações da paciente, de acordo com as normas do Conselho de Revisores Institucionais.
Feminino, 4 anos. Provenientes do Macapá. Criança previamente hígida. É a primogênita do casal, terceira geração de casamento consanguíneo, gestação sem intercorrências. Neurodesenvolvimento normal até os 2 anos. Aos 2 anos e 9 meses, apresentou quadro de febre persistente e regressão do neurodesenvolvimento. Aos 3 anos e 6 meses, iniciou com ocorrências de movimentos clônicos em membros inferiores variáveis, de duração de segundos. Na ocasião passou em consulta com neuropediatra, que questionou a possibilidade de crises epilépticas e iniciou tratamento com valproato de sódio (VPA). Os eventos diminuíram em frequência, mas não cessaram completamente. Aos 3 anos e 10 meses regrediu a afasia e disfagia grave.
Foi admitida em nossas dependências devido essas queixas, além de alteração do nível de consciência. Durante o internamento, realizados exames complementares, de relevância encontramos proteinorraquia e importante alteração em exame de neuroimagem. A RNM crânio e neuroeixo evidenciou alterações de substância branca profunda periventricular, simétricas de substrato inflamatório (Figura 1). Realizado ensaio enzimático para atividade da arilsulfatase A em leucócitos com resultado de 0,90 (VR 5-20), evidenciando enzima insuficiente. Optou-se pela coleta de sequenciamento completo do exoma que evidenciou variante em homozigose no gene ARSA p.Phe377del, diagnóstico compatível com leucodistrofia metacromática (MLD). A menor segue em acompanhamento em nosso serviço para manejo clínico. Foi realizado aconselhamento genético.
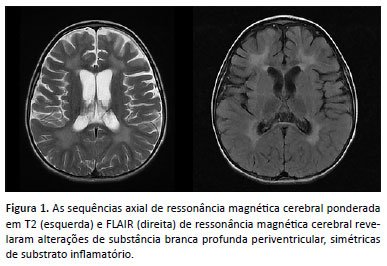
Apresentamos as características clínicas e a avaliação genética de uma paciente brasileira com MLD forma infantil. A MLD é um erro inato do metabolismo lisossomal causado pela deficiência de arilsulfatase A (ARSA), responsável pela hidrólise das ligações éster de galactosil e lactosil sulfatídeos. A baixa concentração dessa enzima provoca o acúmulo intralisossomal em grânulos metacromáticos de sulfatídeo abundante na substância branca do sistema nervoso central (oligodendrócitos, macrófagos e células de Schwann) e do periférico, especialmente nas bainhas de mielina2. O acúmulo de sulfatídeo causa a desmielinização, responsável pela maior parte das manifestações clínicas. Ocorre também formação de depósitos em órgãos viscerais como rins, fígado, vesícula biliar, pâncreas, testículos e córtex adrenal3.
O gene ARSA está localizado no braço longo do cromossomo 22 e consiste em oito éxons, sendo de herança monogênica autossômica recessiva. Já foram relatadas mais de 100 mutações relacionadas. Há uma relação entre genótipo e expressão fenotípica. Pacientes com homozigose para alelos que não expressam nenhuma atividade enzimática apresentam a forma infantil mais grave. Os pacientes que apresentam heterozigose para o alelo nulo evoluem com a forma juvenil ou adulta. Já a homozigose para alelos que expressam baixos níveis da enzima costuma se manifestar como sintomas atenuados na forma adulta4.
A manifestação clínica é dividida em três variantes conforme idade de início dos sintomas. São reconhecidos três tipos de variantes: infantil tardia, entre 12 e 18 meses; juvenil, entre 4 e 16 anos; e início na idade adulta, após a puberdade. Na apresentação precoce predomina o déficit motor, precedendo a deterioração mental, semelhante ao nosso caso.
Em 1932 foram relatados pelo médico Greenfield pela primeira vez os casos de dois pacientes de 3 anos de idade com regressão do neurodesenvolvimento e quadro compatível com leucodistrofia metocromática5.
A variante infantil tardia se manifesta até os 30 meses de vida, mais comumente entre 12 a 24 meses. Caracteriza-se pela rápida e súbita regressão dos marcos do neurodesenvolvimento, principalmente motor, distúrbios de movimentos e arreflexia. Evoluem para tetraplegia espástica, distúrbios de deglutição de tal forma que muitos evoluem com necessidade de gastrostomia. O curso tardio da doença é a evolução para deterioração cognitiva, perda de controle cervical, nistagmo horizontal, atrofia óptica, dor a estímulos leves, distonia, epilepsia e por fim óbito em estado de descerebração1,6.
Por outro lado, a forma juvenil é marcada pelo aparecimento de alterações comportamentais antes dos sintomas motores. Piora no rendimento escolar, comportamentos inadequados e sintomas psiquiátricos têm início insidioso e podem ser erroneamente diagnosticados como esquizofrenia ou depressão. Os sintomas motores, como incoordenação e redução dos reflexos tendinosos, manifestam-se posteriormente. Contudo, uma vez que sintomas neurológicos como hipertonia e espasticidade são estabelecidos, o declínio motor é rápido4,7.
A forma adulta, também de início insidioso, manifesta-se por alterações comportamentais e intelectuais, instabilidade emocional e perda de memória. Ainda que sintomas neurológicos periféricos progressivamente apareçam, a maioria dos pacientes sobrevive por anos6,8.
As alterações em ressonância magnética de crânio já são evidentes logo no início da manifestação dos sintomas. A imagem característica em RNM de crânio no início é de sinal hiperintenso e confluente em corpo caloso e regiões parieto-occipitais e periventriculares. Conforme a doença progride, as lesões avançam para ambos os hemisférios. Outra característica são linhas tigroides na substância branca correspondentes à preservação perivascular de mielina. À espectroscopia, é possível visualizar baixos níveis de N-acetilaspartato, correspondente à perda neuronal difusa, e acúmulo de mio-inositol, compatível com gliose reativa e elevação de colina, referente a aumento do turnover celular da mielina e desmielinização6,7.
O diagnóstico bioquímico é feito com dosagem sérica do nível de atividade da enzima ARSA em leucócitos. Contudo, nos casos de pseudodeficiência, os níveis podem ser baixos e o paciente ser assintomático ou com sintomas brandos. Por outro lado, no caso de níveis normais com manifestação típica e alterações já presentes em RNM, deve-se considerar hipótese de deficiência de saposina B, coativador enzimático essencial da ARSA. Portanto, a dosagem de sulfatídeos em amostra urinária de 24 horas é de extrema importância para complementar a investigação, uma vez que estão significativamente elevados na urina de pacientes com leucodistrofia6,7.
Até o presente momento, não há terapias curativas para a MLD infantil tardia que possam reverter o curso da doença, mas apenas medidas de suporte. O transplante de células tronco hematopoiéticas (TCTH) permite que os monócitos do doador atravessem a barreira hematoencefálica e se diferenciem em micróglia com o objetivo restabelecer os níveis enzimáticos de ARSA. Contudo, a substituição das células da substância branca é um processo lento e pode levar de 12 a 24 meses para a estabilização, ao contrário da rápida progressão da doença. Quanto mais agressivo o fenótipo e quanto maior o intervalo entre início dos sintomas e o transplante, menor a chance de sucesso.
Portanto, o transplante mostrou-se ineficaz para a reversão das lesões e dos sintomas permanentes causados pela doença. Há de se pontuar também que os riscos do transplante, como a evolução para a doença enxerto-hospedeiro, além de que o transplante não reverte as lesões já instaladas no sistema nervoso periférico4,6,7.
Há estudos em andamento para desenvolvimento de terapias alternativas que objetivam modificar o curso natural da doença. A maior dificuldade com a reposição enzimática endovenosa para a MLD é a transposição da barreira hematoencefálica, diferentemente das outras doenças de depósito lisossomal sem envolvimento de sistema nervoso central7,9,10.
Ademais, o TCTH e a terapia gênica são melhores indicadas na fase pré-sintomática ou na fase inicial, períodos de difícil diagnóstico clínico. Na ausência de teste de triagem neonatal, os pacientes são diagnosticados nessas fases somente a partir da identificação de irmão mais velho afetado. Dessa forma, até dois terços dos pacientes são diagnosticados fora da janela de oportunidade. O atraso no diagnóstico pode ser de até 3 anos, enquanto a deterioração nesse período é rápida. Uma metodologia proposta para triagem a partir de sangue seco em papel filtro é a análise escalonada da quantificação de sulfatídeos C 16:0, dosagem da atividade de ARSA e sequenciamento do gene ARSA, com uma especificidade final de 100%9,10.
Uma revisão sistemática aponta que apenas 50% das crianças sobrevivem além de 2,7 anos do aparecimento dos primeiros sintomas e que apenas 25% sobrevivem por até 5 anos. A taxa de sobrevivência está relacionada aos cuidados de suporte (nutrição por gastrostomia, medidas de higiene e uso de antibióticos durante intercorrências infecciosas)7,8.
CONCLUSÃO
A leucodistrofia metacromática é uma doença neurodegenerativa rara e progressiva causada pela deficiência de uma enzima que quebra um lipídio chamado sulfatide. O acúmulo desse no sistema nervoso central causa desmielinização. A doença apresenta-se de três formas, sendo a infantil a mais grave, com início nos primeiros anos de vida e sintomas como regressão do neurodesenvolvimento, problemas de coordenação, convulsões, paralisia e demência. Não existe cura para a MLD, mas o diagnóstico e tratamento precoces podem ajudar a retardar a progressão da doença. O tratamento geralmente inclui fisioterapia, terapia ocupacional e medicamentos para controlar os sintomas.
REFERÊNCIAS
1. Espejo LM, de la Espriella R, Hernández JF. Leucodistrofia metacromática. Presentación de caso. Rev Colomb Psiquiatr. 2017;46(1):44-9. DOI: 10.1016/j.rcp.2016.05.001
2. Alvarez-Pabón Y, Lozano-Jiménez JF, Di Lizio-Miele KG, Contreras-García GA. Late infantile metachromatic leukodystrophy: case report. Arch Argent Pediatr. 2019;117(1):e52-e5.
3. Alencar B, Carvalho T. Leucodistrofia Metacromática Infantil: principais manifestações clínicas e a atuação da fisioterapia. Rio de Janeiro: Anima Educação; 2023 [citado 26 set 2024]. Disponível em: https://repositorio.animaeducacao.com.br/items/2e2c6243-a76e-4318-8e52-89af417201ac
4. Biffi A, Lucchini G, Rovelli A, Sessa M. Metachromatic leukodystrophy: an overview of current and prospective treatments. Bone Marrow Transplant. 2008;42(Suppl 2):S2-6.
5. Greenfield JG. A Form of Progressive Cerebral Sclerosis in Infants associated with Primary Degeneration of the Interfascicular Glia. Proc R Soc Med. 1933;26(6):690-7.
6. Gieselmann V, Krägeloh-Mann I. Metachromatic leukodystrophy--an update. Neuropediatrics. 2010;41(1):1-6.
7. van Rappard DF, Boelens JJ, Wolf NI. Metachromatic leukodystrophy: Disease spectrum and approaches for treatment. Best Pract Res Clin Endocrinol Metab. 2015;29(2):261-73. DOI: 10.1016/j.beem.2014.10.001
8. Mahmood A, Berry J, Wenger DA, Escolar M, Sobeih M, Raymond G, et al. Metachromatic leukodystrophy: a case of triplets with the late infantile variant and a systematic review of the literature. J Child Neurol. 2010;25(5):572-80.
9. Laugwitz L, Schoenmakers DH, Adang LA, Beck-Woedl S, Bergner C, Bernard G, et al. Newborn screening in metachromatic leukodystrophy - European consensus-based recommendations on clinical management. Eur J Paediatr Neurol. 2024;49:141-54.
10. Morton G, Thomas S, Roberts P, Clark V, Imrie J, Morrison A. The importance of early diagnosis and views on newborn screening in metachromatic leukodystrophy: results of a Caregiver Survey in the UK and Republic of Ireland. Orphanet J Rare Dis. 2022;17(1):403.
Hospital Pequeno Príncipe, Neurologia infantil - Curitiba - PR - Brasil
Endereço para correspondência:
Lisandra Coneglian de Farias
Hospital Pequeno Príncipe
Rua Desembargador Motta, nº 1070, Água Verde
Curitiba, PR, Brasil. CEP: 80250-060
E-mail: lisandra.coneglian@gmail.com
Data de Recebimento: 15/04/2024
Data de Aprovação: 30/09/2024
