
INTRODUÇÃO
Erros inatos do metabolismo representam distúrbios hereditários resultantes de mutações genéticas que afetam as enzimas ou proteínas envolvidas nas vias metabólicas. Essas mutações levam a falhas no processamento de nutrientes, produção de energia ou eliminação de resíduos metabólicos, refletindo no acúmulo de substâncias tóxicas ou na falta de produtos metabólicos essenciais1. Um exemplo notável de erro metabólico é a fenilcetonúria (PKU), uma doença autossômica recessiva originada por mutações na enzima fenilalanina-hidroxilase (PAH), resultando em um déficit no metabolismo do aminoácido fenilalanina (Phe). Consequentemente, ocorre um acúmulo de fenilalanina no plasma, ocasionando efeitos prejudiciais em múltiplos sistemas do corpo, incluindo o sistema nervoso, pele e composição da urina1,2.
Os dados epidemiológicos relacionados à fenilcetonúria podem variar na literatura, devido a diferentes fatores, como variações genéticas, cobertura de triagem neonatal e abordagens metodológicas adotadas nos estudos. No Brasil, segundo o Ministério da Saúde, tem sido encontrada uma prevalência variando de 1:15.000 a 1:25.000 nascidos vivos3. Um estudo abordou a triagem neonatal para PKU em Sergipe, Brasil, relatando 1 caso a cada 8.690 indivíduos, demonstrou que apesar da cobertura adequada e diagnóstico precoce, ocorreram atrasos no tratamento e desafios no controle metabólico4.
O diagnóstico precoce e a intervenção dietética são componentes cruciais na prevenção de danos cerebrais irreversíveis decorrentes da fenilcetonúria2. Uma revisão sistemática abordou o impacto da PKU na América Latina. Os autores identificaram uma prevalência estimada de 18% para deficiência intelectual, 15% para atraso motor e 35% para déficit de fala em pacientes diagnosticados precocemente. Adicionalmente, essa análise identificou lacunas nas necessidades neuropsicológicas e desafios socioeconômicos que não foram adequadamente tratados, ressaltando a importância de direcionar intervenções para abordar essas questões complexas e fornecer melhorias substanciais5.
A fenilcetonúria impõe um impacto significativo na vida e na alimentação dos indivíduos afetados, devido ao tratamento restritivo e vitalício necessário. A abordagem terapêutica envolve a adoção de uma dieta de restrição proteica, que inclui a redução do consumo de proteínas naturais, complementada com misturas especiais de aminoácidos. O principal objetivo dessa intervenção é garantir o equilíbrio metabólico, ao mesmo tempo em que fornece a energia e os nutrientes necessários para o crescimento adequado6. A falta de aderência rigorosa à dieta, aliada à natureza restritiva desta, pode resultar em diversas complicações, especialmente em crianças e adolescentes que estão desenvolvendo autonomia nas escolhas alimentares3.
Apesar dos progressos recentes nas pesquisas sobre PKU, persistem inconsistências na literatura, especialmente no que tange a medidas antropométricas e parâmetros bioquímicos, que influenciam o risco de doenças cardiovasculares nesses pacientes. Evidências atuais indicam que, apesar dos avanços na terapia dietética, resultados de crescimento considerados ideais ainda não são alcançados na PKU7. Portanto, o objetivo deste estudo é investigar os parâmetros antropométricos, metabólicos e os níveis séricos de fenilalanina em pacientes com fenilcetonúria.
MATERIAIS E MÉTODOS
Trata-se de um estudo de coorte, realizado em um ambulatório de referência no atendimento de crianças com fenilcetonúria no estado de Sergipe. O serviço é vinculado ao Sistema Único de Saúde (SUS) e é responsável pela busca ativa de casos suspeitos da doença, confirmação diagnóstica, tratamento e acompanhamento multidisciplinar especializado.
Os critérios de inclusão foram: pacientes com diagnóstico de fenilcetonúria, com idade entre 4 e 21 anos, acompanhados no referido serviço no período de janeiro de 2017 a janeiro de 2020. Foram excluídos pacientes com diagnóstico tardio de PKU, doenças osteometabólicas crônicas, doença renal crônica prévia ou uso de medicamentos que pudessem interferir nos resultados dos exames bioquímicos.
As medidas antropométricas foram realizadas pela pediatra responsável do ambulatório, numa frequência aproximada de 4 meses. O peso foi registrado em quilogramas (kg), aferido em balança eletrônica, com precisão de 100 gramas (g). A estatura foi medida em centímetros (cm), utilizando o estadiômetro vertical, com precisão de milímetros (mm). O Índice de Massa Corporal (IMC) foi calculado utilizando a fórmula IMC = peso (kg) / altura ao quadrado (m²) e os valores foram avaliados de acordo com os percentis da curva de crescimento da Organização Mundial de Saúde (OMS).
Os exames laboratoriais foram realizados no laboratório de análises clínicas do próprio hospital. Foram coletadas amostras de sangue para dosagens de fenilalanina (Phe) a cada 4 meses, além de coletas anuais para análise de triglicerídeos, colesterol total e frações, glicemia de jejum e insulina basal, além de marcadores de metabolismo ósseo. As amostras foram colhidas em tubos adequados, com jejum de 10 a 14 horas, e os exames foram realizados por métodos enzimáticos colorimétricos e de rotina laboratorial. Os valores de corte de fenilalanina utilizados foram entre 2 e 6mg/dL em todas as idades, conforme Vockley et al. (2014)8.
Os dados foram coletados através dos registros em prontuários. Após a coleta, foram transferidos para uma planilha no Excel for Windows 10 e foram realizadas medidas descritivas e análises comparativas. A normalidade dos dados foi verificada utilizando o teste de Kolmogorov-Smirnov. Para associação entre as variáveis, foi aplicado o teste de Fisher, e para comparação entre as variáveis, utilizaram-se os testes t não pareado e Kruskal-Wallis. O nível de significância adotado foi p<0,05.
O estudo recebeu aprovação do Comitê de Ética em Pesquisa (número do parecer 3676244, CAAE 17507219.6.0000.5546), e os participantes foram convidados a assinar o Termo de Consentimento Livre e Esclarecido (TCLE) e/ou termo de Assentimento. Foram adotadas medidas para garantir a confidencialidade e anonimato dos participantes.
RESULTADOS
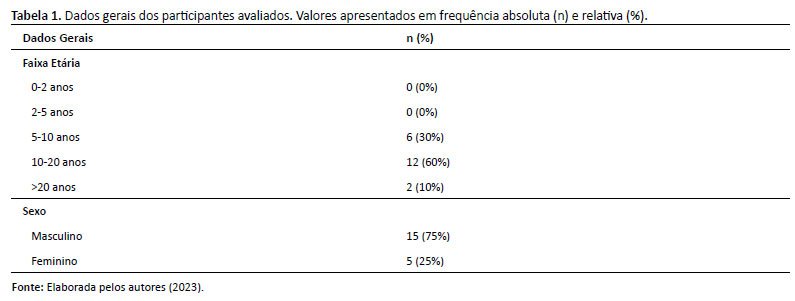
Após a aplicação dos critérios de inclusão e exclusão, uma amostra de 20 pacientes foi obtida, sendo a maioria (75%) do sexo masculino. A idade média da amostra foi de 13,26 anos (± 3,98) e mediana de 12,85, com 30% dos pacientes na faixa etária de 6 a 10 anos e 70% na faixa etária de 10 a 20 anos. Os dados gerais dos participantes são apresentados na Tabela 1.
Na análise antropométrica, foram realizadas oito medidas de peso, altura e IMC para cada paciente. A maioria dos pacientes apresentou peso, altura e IMC dentro dos parâmetros esperados, com médias de 33,76kg (± 14,55), 125cm (± 20,48) e 16,71kg/m2 (± 3,56), respectivamente.
Em relação ao peso, os pacientes foram avaliados e classificados em três grupos: peso adequado para idade, peso elevado para idade e baixo peso para idade. Das medidas obtidas, a maioria apresentou peso adequado para a idade. Comparando entre os sexos, a média de peso obtida para os indivíduos do sexo masculino foi 33,82 (DP ± 16,19), já para os indivíduos do sexo feminino, a média obtida foi de 33,57 (DP ± 9,36).
Quanto à altura, os pacientes foram classificados como “altura adequada para idade” ou “baixa estatura para a idade”, de forma que a maioria apresentou medidas de altura dentro do esperado. A média da altura obtida para os indivíduos do sexo masculino foi 125,9 (DP ± 20,48), ao passo que para os indivíduos do sexo feminino, a média obtida foi de 122,3 (DP ± 22,61).
A análise do IMC foi considerada de acordo com o último parâmetro antropométrico avaliado, o qual foi classificado em eutrófico, sobrepeso, obesidade, magreza e magreza extrema. A maioria dos pacientes foi classificada como eutrófica. Os pacientes com IMC elevado (sobrepeso ou obesidade) corresponderam a 25%, ao passo que aqueles com baixo IMC apresentaram 10% dos participantes. A média do IMC foi de 21,61kg/m2 (± 1,97) para os indivíduos eutróficos, 18,1kg/m2 (± 0,31) para os indivíduos com magreza e 14,47kg/m2 (± 1,64) para os indivíduos com magreza extrema.
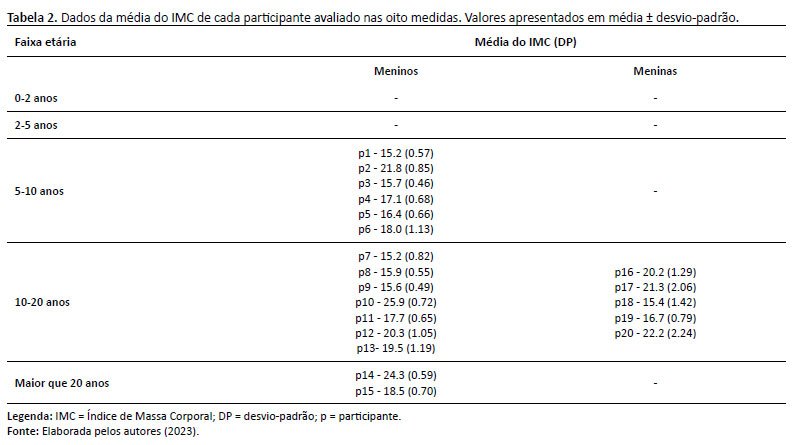
A média do IMC obtida para os indivíduos do sexo masculino foi 16,89 (DP ± 3,69), enquanto para os indivíduos do sexo feminino a média obtida foi de 16,17 (DP ± 3,49). A tabela 2 apresenta a média do IMC de cada paciente durante os oito momentos avaliados, conforme o sexo e a idade.
Analisando o IMC em 2 momentos, observou-se um total de 15 (75%) indivíduos com o IMC adequado na primeira medida do estudo, chegando a reduzir para um total de 11 (55%) eutróficos na última medida realizada. Os pacientes com IMC acima do esperado no início da coleta corresponderam a um total de 3 (15%), sendo 1 (5%) com sobrepeso e 2 (10%) com obesidade, passando para 5 (25%) ao final da coleta, sendo 3 (15%) com sobrepeso e 2 (10%) obesos. O resultado para magreza e magreza extrema foi de 1 (5%) para ambas ao início, passando para 2 (10%) pacientes com magreza e nenhum com magreza extrema ao final do período. Analisando a estatura, percebeu-se adequada estatura para idade em 19 (95%) e 14 (70%) pacientes no início e final do estudo, respectivamente, havendo 1 (5%) e 3 (15%) pacientes abaixo da altura desejada nos mesmos períodos.
Foram realizados exames laboratoriais anualmente para avaliar o perfil metabólico dos pacientes. A média final dos valores dos exames foi calculada. A média dos valores absolutos dos exames foi: glicemia 65,25mg/dL (± 23,36), aspartato aminotransferase (AST) 21,32U/L (± 10,27), alanina aminotransferase (ALT) 17,17U/L (± 8,57), LDL 56,10mg/dL (± 24,22), HDL 27,75mg/dL (± 11,84), colesterol total 50,60mg/dL (± 45,53), triglicerídeos 56,85mg/dL (± 36,36), cálcio total 7,95mg/dL (± 2,83), cálcio ionizado 3,42mg/dL (± 1,71), albumina 3,29g/dL (± 1,35) e fósforo 3,44mg/dL (± 1,66).
Destaca-se, dentre os exames acompanhados, níveis de HDL majoritariamente baixos, chegando a corresponder a 90% do total de pacientes na média dos valores durante as coletas analisadas. Do mesmo modo, os valores médios de cálcio e cálcio ionizado estiverem abaixo do esperado em 40% e 50%, respectivamente, dos pacientes nas amostras em questão.
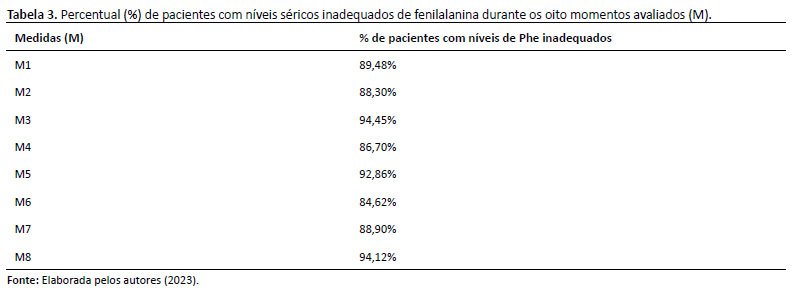
A dosagem sérica de fenilalanina foi feita a cada quatro meses, sendo realizadas oito medidas séricas de fenilalanina para cada paciente. A média geral de fenilalanina foi de 11,5mg/dL (± 3,68). A maioria dos pacientes apresentou níveis séricos inadequados de fenilalanina durante o período de acompanhamento, conforme a tabela 3, a seguir:
DISCUSSÃO
A associação entre sobrepeso e obesidade em indivíduos com fenilcetonúria (PKU) tem sido objeto de debate na literatura. Na presente pesquisa, aproximadamente um quarto dos pacientes avaliados apresentou IMC elevado em sua última visita. Em uma revisão sistemática recente, os autores observaram que o excesso de peso foi um evento frequente nas crianças e adolescentes com PKU, especialmente após os oito anos de vida. Dentre os fatores associados, destacaram-se o maior consumo de alimentos caloricamente densos devido à restrição proteica e a falta de estímulos para a prática de atividades físicas decorrente da retração social4,9. Em um estudo transversal com 94 adolescentes portadores de PKU, foram investigadas diversas variáveis, incluindo o percentual de gordura corporal, o IMC e o controle metabólico. Os resultados revelaram que fatores como gênero feminino, excesso de peso e maior consumo de proteínas estavam associados a níveis mais elevados de gordura corporal9.
Ressalta-se que, embora o IMC seja amplamente utilizado como medida de resultado do crescimento, ele possui algumas limitações, visto que a contribuição relativa da massa magra e gorda para o peso corporal não pode ser avaliada. Com base apenas no IMC, alguns pacientes podem parecer relativamente estáveis em termos de peso, apesar de mudarem em termos de relação gordura/lean subjacente10. Nesse sentido, a circunferência da cintura possui correlação com parâmetros relacionados ao risco cardiovascular nos pacientes com PKU, sugerindo que esta pode ser um indicador mais preciso de risco cardiovascular quando comparado ao IMC11.
A maioria dos pacientes deste estudo apresentou estatura adequada para a idade. Semelhantemente, um estudo revisou o crescimento de crianças com PKU, concluindo que esse parâmetro obteve resultados normais ao nascimento e durante a infância, apesar de haver uma estatura significativamente menor nos primeiros quatro anos de vida em comparação com populações de referência. Em contraste, pacientes com hiperfenilalaninemia leve, que não requerem restrição dietética, não apresentaram comprometimento do crescimento6.
Em relação ao perfil lipídico, a análise dos exames laboratoriais identificou que os indivíduos apresentaram valores médios de LDL, triglicerídeos e colesterol total dentro da faixa esperada para a idade. Entretanto, o nível médio de HDL foi considerado baixo em comparação com a idade da amostra avaliada. De maneira semelhante, o estudo de Almeida et al. (2020)12 demonstrou que a maioria dos pacientes apresentou adequação de acordo com os parâmetros antropométricos e testes bioquímicos, exceto pelos níveis mais inferiores de HDL em 59,3% dos pacientes. Considerando que, além dos parâmetros clínicos, baixos níveis de HDL e níveis elevados de LDL isoladamente foram identificados como indicadores favoráveis de risco cardiovascular12, pacientes com PKU necessitam de uma atenção quanto ao monitoramento clínico e laboratorial para prevenir alterações metabólicas, além de evitar o ganho excessivo de peso e suas implicações.
A dieta desempenha um papel fundamental no manejo da fenilcetonúria e o consumo de carboidratos pode influenciar o controle glicêmico. No entanto, neste estudo, não foram observados níveis elevados de glicose no sangue dos indivíduos monitorados, o que difere das sugestões da literatura, cujas restrições dietéticas podem resultar em níveis glicêmicos mais altos devido ao aumento da ingestão calórica9. A restrição de proteínas naturais favorece, e pode até estimular fisiologicamente, o consumo de alimentos ricos em carboidratos, especialmente carboidratos simples, levando a uma dieta de índice glicêmico mais alto e aumentando a ocorrência de anormalidades metabólicas e excesso de peso3,13.
Apesar dos riscos associados à alta ingestão de carboidratos na literatura, poucas pesquisas foram realizadas sobre o metabolismo de carboidratos em pacientes com PKU. Em um estudo multicêntrico, Couce et al. (2018)9 avaliaram um total de 83 pacientes com idade entre 4 e 52 anos e identificaram que o controle metabólico foi adequado em 71,9%. Os autores perceberam que os pacientes com maior risco de intolerância a carboidratos e resistência à insulina eram adultos e com excesso de peso.
Pacientes com fenilcetonúria também podem apresentar um risco aumentado de deficiências de micronutrientes14. Observou-se, neste estudo, que os níveis médios de cálcio e cálcio ionizado apresentaram uma discreta redução neste mineral. Além disso, o nível médio de fósforo também foi considerado baixo, suscitando a dúvida se esses pacientes estavam aderindo adequadamente à dieta prescrita. Salienta-se que no grupo de crianças com PKU, tal como na população global, a avaliação da saúde óssea deve ser realizada por densitometria óssea15. A relação entre PKU e risco de doenças ósseas é reconhecida, porém a eficácia da suplementação de cálcio como prevenção ainda carece de evidências sólidas e não é amplamente adotada na prática clínica16.
O controle preciso dos níveis de Phe no sangue é crucial para prevenir problemas cognitivos, neuropsicológicos e comportamentais associados à fenilcetonúria. A categorização da doença em fenótipos, com base nos níveis prévios de Phe, como PKU clássica, leve e hiperfenilalaninemia, permite antecipar complicações como microcefalia em crianças e questões neurológicas como Transtorno do Déficit de Atenção com Hiperatividade (TDAH) em diferentes estágios da vida. Ademais, a monitorização dos níveis séricos de fenilalanina é essencial para orientar decisões clínicas, como ajustes na dieta ou terapia de substituição enzimática, visando ao melhor controle da doença17.
Neste estudo, a abordagem com 20 pacientes com fenilcetonúria destaca a complexidade em alcançar níveis normais de fenilalanina durante o acompanhamento, evidenciando uma maior necessidade do aumento da conscientização sobre o tema. Essa situação foi constatada tanto na média final quanto em medidas isoladas, resultando em níveis elevados para todos os pacientes. Estudos demonstram que o controle dietético na fenilcetonúria tende a diminuir em crianças maiores e adolescentes, com dificuldades relatadas pelos pais devido à entrada das crianças na escola, resultando em alterações nas dosagens de fenilalanina após os 10 anos18,19. Outro fator que pode justificar os desajustes nos níveis de Phe encontra-se na dificuldade de acesso e acompanhamento de profissionais especializados, de forma que a ausência de um sistema de referência para os serviços de PKU pode contribuir com a ausência de orientação dos pacientes sobre a relevância da dieta20.
Uma limitação importante deste estudo é a falta de avaliação dietética, essencial para monitorar os pacientes adequadamente, juntamente com a falta de acompanhamento contínuo e registros detalhados nos prontuários. Além disso, a complexidade de comparar resultados com outros estudos de PKU devido a diferenças nas idades dos pacientes e faixas etárias adotadas nas metodologias ressalta a necessidade de futuras pesquisas, incluindo avaliações de longo prazo sobre os efeitos da restrição de fenilalanina e a adesão à dieta.
Em síntese, este estudo avaliou parâmetros antropométricos, metabólicos e níveis de fenilalanina em pacientes com fenilcetonúria. A maioria dos pacientes apresentou estado nutricional adequado, apesar das alterações nos níveis baixos de HDL, e inadequado controle dos níveis séricos de fenilalanina. Os achados ressaltam a necessidade de abordagem mais abrangente para otimizar o controle metabólico, destacando a importância do monitoramento constante e intervenção dietética, com acompanhamento multidisciplinar, para prevenir efeitos adversos e melhorar a qualidade de vida dos pacientes com fenilcetonúria.
REFERÊNCIAS
1. Schiergens KA, Weiß KJ, Dokoupil K, Fleissner S, Maier EM. Ernährung bei angeborenen Stoffwechselerkrankungen – ein Spagat zwischen Genuss und Therapie. Bundesgesundheitsbl. 2020;63(Suppl 2):864-71. DOI: https://doi.org/10.1007/s00103-020-03168-x.
2. Kanufre VC, Soares RDL, Alves MRA, Aguiar MJB, Starling ALP, Norton RC. Metabolic syndrome in children and adolescents with phenylketonuria. J Pediatr (Rio J). 2015;91(1):98-103. DOI: https://doi.org/10.1016/j.jped.2014.06.006.
3. Tummolo A, Carella R, Paterno G, Bartolomeo N, Giotta M, Dicintio A, et al. Body composition in adolescent PKU patients: beyond fat mass. Children (Basel). 2022;9(9):1353. DOI: https://doi.org/10.3390/children9091353.
4. Camatta GC, Kanufre VDC, Alves MRA, Soares RDL, Norton RC, Aguiar MJB, et al. Body fat percentage in adolescents with phenylketonuria and associated factors. Mol Genet Metab Rep. 2020;23:100595. DOI: https://doi.org/10.1016/j.ymgmr.2020.100595.
5. Sena BS, Andrade MIS, Silva APF, Dourado KF, Silva ALF. Excesso de peso e fatores associados em crianças e adolescentes com fenilcetonúria: uma revisão sistemática. Rev Paul Pediatr. 2020;38:e2018201.
6. Shakiba M, Alaei M, Saneifard H, Mosallanejad A. Avaliação de índices antropométricos em pacientes com fenilcetonúria. Iran J Child Neurol. 2020;14(2):27.
7. Fernandez-Crespo S, Vazquez-Agra N, Marques-Afonso AT, Cruces-Sande A, Martinez-Olmos MA, Araujo-Vilar D, et al. The value of waist circumference as a predictor of cardiovascular risk in adult patients with classic phenylketonuria. Med Clin (Barc). 2023. DOI: https://doi.org/10.1016/j.medcli.2023.06.023.
8. Vockley J, Andersson HC, Antshel KM, Braverman NE, Burton BK, Frazier DM, et al. Phenylalanine hydroxylase deficiency: diagnosis and management guideline. Genet Med. 2014;16(2):188-200. DOI: https://doi.org/10.1038/gim.2013.157.
9. Couce ML, Sánchez-Pintos P, Vitoria I, De Castro MJ, Aldámiz-Echevarría L, Correcher P, et al. Carbohydrate status in patients with phenylketonuria. Orphanet J Rare Dis. 2018;13(1):103. DOI: https://doi.org/10.1186/s13023-018-0847-x.
10. Green B, Browne R, Firman S, Hill M, Rahman Y, Hansen KK, et al. Nutritional and metabolic characteristics of UK adult phenylketonuria patients with varying dietary adherence. Nutrients. 2019;11(10):1-13. DOI: https://doi.org/10.3390/nu11102459
11. Alghamdi N, Alfheeaid H, Cochrane B, Adam S, Galloway P, Cozens A, et al. Mechanisms of obesity in children and adults with phenylketonuria on contemporary treatment. Clin Nutr ESPEN. 2021;46:539-43. DOI: https://doi.org/10.1016/j.clnesp.2021.10.012.
12. de Almeida BN de F, Laufer JA, Mezzomo TR, Shimada NC, Furtado IHF, Dias MRMG, et al. Parâmetros nutricionais e metabólicos de crianças e adolescentes com fenilcetonúria. Clin Nutr ESPEN. 2020;37:44-9.
13. Moretti F, Pellegrini N, Salvatici E, Rovelli V, Banderali G, Radaelli G, et al. Dietary glycemic index, glycemic load and metabolic profile in children with phenylketonuria. Nutr Metab Cardiovasc Dis. 2017;27(2):176-82. DOI: https://doi.org/10.1016/j.numecd.2016.11.002.
14. Kose E, Arslan N. Vitamin/mineral and micronutrient status in patients with classical phenylketonuria. Clin Nutr. 2019;38(1):197-203. DOI: https://doi.org/10.1016/j.clnu.2018.01.034.
15. van Wegberg AMJ, MacDonald A, Ahring K, Belanger-Quintana A, Blau N, Bosch AM, et al. The complete European guidelines on phenylketonuria: diagnosis and treatment. Orphanet J Rare Dis. 2017;12(1):162. DOI: https://doi.org/10.1186/s13023-017-0685-2.
16. Tanaka NYY, Turcato MF, Nicoletti CF, Nonino CB, Martins LD, Iannetta O, et al. Effects of short-term calcium supplementation in children and adolescents with phenylketonuria. J Clin Densitom. 2018;21(1):48-53. DOI: https://doi.org/10.1016/j.jocd.2017.02.001.
17. Silveira AM. Sobrepeso/obesidade e doença hepática gordurosa não alcoólica em adolescentes com fenilcetonúria [tese]. Belo Horizonte: Universidade; 2020. Disponível em: https://pesquisa.bvsalud.org/portal/resource/pt/biblio-1397300.
18. Rovelli V, Longo N. Phenylketonuria and the brain. Mol Genet Metab. 2023;139(1):107583. DOI: https://doi.org/10.1016/j.ymgme.2023.107583.
19. Karam SM, Jardim LB, Giugliani R, Horta BL. Triagem neonatal para hiperfenilalaninemia: um estudo de coorte. Rev AMRIGS. 2012;56(1):17-21.
20. Lamônica DA, Gejão MG, Anastácio-Pessan FL. Fenilcetonúria e habilidades de leitura e escrita. Rev CEFAC. 2015;17(1):143-50. DOI: https://doi.org/10.1590/1982-0216201515313.
Data de Recebimento: 06/01/2024
Data de Revisão: 06/01/2024
Data de Aprovação: 14/01/2024
Data de Publicação: 01/07/2025
