A síndrome de Cornélia de Lange (SCdL) é uma doença rara, caracterizada por múltiplas malformações, como atraso no crescimento psicomotor, alterações cardíacas, gastrointestinais, musculoesquelética e fácies típicas1. Decorre de mutações em genes que estruturam ou regulam o complexo da coesina, que, por sua vez, tem papel na coesão cromatídica, reparo do DNA e expressão gênica2.
A prevalência varia de 1:10.000 a 1:30.000 nascidos vivos. Devido à heterogeneidade no fenótipo, o distúrbio pode se apresentar de diferentes formas, o que dificulta o diagnóstico3. A etiologia é ainda incerta em muitos pacientes e o risco de recorrência é de 2 a 5% para irmãos de um indivíduo afetado4.
O diagnóstico, além de se basear em critérios clínicos, pode ser evidenciado através da detecção de cópias anormais e/ou variação da sequência do genótipo. No pré-natal o diagnóstico pode ser desafiador, previsto a partir da junção de características identificadas na ultrassonografia (USG), como aumento da translucência nucal, crescimento intrauterino restrito (CIUR) simétrico, anomalias dos membros e face.
Este relato de caso tem como objetivo correlacionar achados e condutas com dados da literatura.
RELATO DE CASO
Recém-nascido (RN) a termo, nascido de parto cesárea, com 37 semanas e 5 dias de idade gestacional, Apgar 9/10, sem necessidade de reanimação neonatal, peso 1.935g (pequeno para a idade gestacional). Perímetro cefálico de 31cm (microcefalia), perímetro abdominal de 29cm, perímetro torácico de 29cm e comprimento de 44cm ao nascer.
Mãe de 23 anos, G2PN1A0, apresentou histórico de diabetes gestacional. Suscetível à toxoplasmose; VDRL e HBsAg foram não reatores. Condiloma acuminado com 24 semanas de idade gestacional. Malformações fetais já presumidas em ultrassonografias durante o pré-natal. Realizada USG com idade gestacional de 12 semanas e 1 dia mostrou translucência nucal de 3,2mm (valor de referência – VR: <2,5mm); USG morfológica com 24 semanas revelou mesomelia, acromelia e oligodactilia bilateral; USG com 35 semanas e 5 dias indicou peso fetal inferior ao percentil 3 (p3), com circunferência abdominal também abaixo do p3. Sem contexto infeccioso na gestação e período periparto.
Ao nascimento foram constatadas as seguintes malformações: orelhas com baixa implantação, fronte proeminente, retrognatismo, acromegalia, oligodactilia bilateral, membros superiores curtos, genitália ambígua (ausência de testículos em bolsa escrotal, micropênis), tufo de pelos na região sacral (Figuras 1 e 2). A partir das características fenotípicas, com avaliação pormenorizada das características faciais e o auxílio de geneticista, levantou-se a hipótese diagnóstica de SCdL, confirmada pelos critérios clínicos preconizados pelo Primeiro Consenso Internacional de Diagnóstico e Manejo da SCdL3.
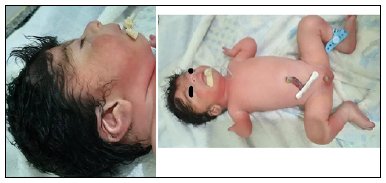 Figura 1. Baixa implantação das orelhas; filtro nasal longo; comissuras labiais com inclinação para baixo; encurtamento de membros superiores; oligodactilia bilateral.
Figura 1. Baixa implantação das orelhas; filtro nasal longo; comissuras labiais com inclinação para baixo; encurtamento de membros superiores; oligodactilia bilateral. Figura 2. Genitália ambígua; oligodactilia; hipertelorismo na região sacral.
Figura 2. Genitália ambígua; oligodactilia; hipertelorismo na região sacral.Ecocardiograma mostrou comunicação interatrial (CIA) tipo ostium secundum de 1,42mm, com shunt esquerda-direita e relação Qp/Qs estimada de 1,06. Avaliação tomográfica do crânio apontou alargamento do espaço subaracnoide em fossa média e fissuras silvianas, cavum do septo pelúcido, ausência de sinais de processos expansivos e sistema ventricular com topografia, morfologia e dimensões normais. USG da região inguinal evidenciou presença de testículos. USG de rins e vias urinárias não exibiu alterações.
Inicialmente o RN foi encaminhado para alojamento conjunto. Foi necessário o uso de sonda orogástrica (SOG) para nutrição, em virtude de dificuldade de sucção. Após administração de dieta via SOG, o RN apresentou episódios de vômitos em grande quantidade, sendo então encaminhado à UTI neonatal. Apresentou crise convulsiva, recebendo fenobarbital (20mg/kg via SOG) e episódio de hipertermia (39,1ºC). Foi mantido fenobarbital em dose de manutenção. Para diagnóstico diferencial com hiperplasia adrenal congênita, foram solicitados os seguintes exames: 17-OH-progesterona: 1628mcg/dL (VR: até 22); testosterona total: 147ng/dL (VR adultos: 165-173); androstenediona: 6,1ng/mL (VR: <1,6); cortisol basal: 9,5mcg/dL (VR: 5,3-22,5); sódio: 134mEq/dL; potássio: 6,7mEq/dL.
Cursou com baixo aporte hídrico por SOG e oligúria. Tentativas iniciais de obtenção de acesso central de inserção periférica (PICC) foram mal sucedidas. Recebeu terapia de reidratação via SOG, com melhora da diurese. Evoluiu com tolerância à dieta via SOG, mas não progrediu na sucção/deglutição, sendo necessária a realização de GTT. Com boa tolerância à dieta por GTT e adequado ganho ponderal, recebeu alta hospitalar com 63 dias de vida para acompanhamento ambulatorial.
DISCUSSÃO
A SCdL é uma síndrome rara, que afeta o crescimento, o comportamento e o desenvolvimento cognitivo, além de apresentar manifestações sistêmicas3,5-7. O tipo clássico pode ser reconhecido clinicamente por meio das características físicas da criança3. Na variante não clássica a criança pode apresentar características mais discretas8; essa variante representa a maioria dos casos9.
O diagnóstico pré-natal pode ser realizado, todavia, é raro. Alterações ultrassonográficas podem auxiliar na hipótese diagnóstica, como aumento da translucência nucal, CIUR simétrico, malformações em membros e anomalias em face1,10-13. Estudo molecular pode ser realizado por amniocentese, auxiliando no diagnóstico. Contudo, caso não sejam encontradas mutações, não é possível excluir o diagnóstico1,3.
No caso relatado os seguintes exames pré-natais mostraram:
1. USG com IG de 12 semanas e 1 dia: translucência nucal = 3,2mm;
2. USG morfológica com IG de 24 semanas: mesomelia, acromelia e oligodactilia bilateral;
3. USG com IG de 35 semanas e 5 dias: peso fetal < p3, circunferência abdominal < p3.
É evidente que o feto apresentava características que poderiam incluir a SCdL nos principais diagnósticos diferenciais. No entanto, não foi solicitado teste genético, haja vista que o resultado não iria interferir na conduta adotada posteriormente, um resultado negativo não excluiria a hipótese e o custo seria elevado.
O diagnóstico pós-natal de crianças com malformações exige exame físico minucioso14. Na SCdL, o diagnóstico pode ser estabelecido por meio de critérios clínicos e/ou por rastreamento genético com mutações sugestivas de SCdL, envolvendo os genes NIPBL, SMC1A, SMC3, RAD21, BRD4, HDAC8 e ANKRD113,8.
O Primeiro Consenso Internacional de Diagnóstico e Manejo da SCdL estabeleceu em 2018 os seguintes critérios diagnósticos3:
Sinais cardinais (2 pontos)
1. Encontro das sobrancelhas na linha média facial e/ou sobrancelhas grossas;
2. Nariz curto, nariz côncavo e/ou ponta nasal antevertida;
3. Buço longo e/ou sem sulco;
4. Lábio superior vermelho e/ou comissura labial voltada para baixo;
5. Oligodactilia nas mãos ou mãos com ausência de dedos;
6. Hérnia diafragmática.
Sinais sugestivos (1 ponto)
1. Déficit no desenvolvimento e/ou déficit cognitivo;
2. Déficit no crescimento pré-natal;
3. Déficit no crescimento pós-natal;
4. Microcefalia;
5. Mãos pequenas e/ou pés;
6. Quinto dedo pequeno;
7. Hirsutismo.
O diagnóstico de SCdL clássica é definido por uma pontuação maior ou igual a 11, estando presentes pelo menos 3 sinais cardinais. De 9 a 10 pontos, com pelo menos 2 sinais cardinais, o diagnóstico de SCdL não clássica é estabelecido. Se 4 a 8 pontos com pelo menos 1 sinal cardinal, está indicado teste molecular para a confirmação do diagnóstico. Caso a pontuação seja menor que 4 pontos não se indica o teste molecular3.
O paciente do relato apresentava: encontro das sobrancelhas na linha média e sobrancelhas grossas, nariz curto e côncavo, buço longo e sem sulco, oligodactilia nas mãos, déficit de crescimento pré-natal, microcefalia, mãos pequenas e hirsutismo, totalizando 14 pontos; foi diagnosticada SCdL sem a necessidade de um exame molecular.
Após diagnóstico da síndrome é necessário rastrear a criança para malformações; solicita-se ecocardiograma, em função da alta prevalência de cardiopatias congênitas, e USG renal. Exames de imagem do sistema nervoso central estão indicados na presença de convulsões3.
No caso relatado o ecocardiograma evidenciou CIA sem repercussão. USG de rins e vias urinárias foi normal. Testículos foram localizados na região inguinal. TC de crânio, apesar de não estar formalmente indicada no momento do diagnóstico, foi importante para afastar outras causas de múltiplas malformações.
Existem curvas de crescimento e peso específicas para o acompanhamento de crianças portadoras da SCdL, que devem ser utilizadas pelo pediatra que realizará a puericultura8,15, o qual também deverá orientar aos pais que a vacinação segue o calendário vacinal usual3.
A dificuldade de alimentação está presente em muitos casos, podendo ter várias causas, como malformações gastrointestinais e dificuldade de sucção, sendo necessária abordagem multidisciplinar, com participação de fonoaudiólogo3,8. Sempre que possível deve-se manter a alimentação oral; no entanto, em alguns casos a GTT pode ser necessária2,3,8.
CONCLUSÃO
Tendo em vista que a SCdL tem uma incidência rara, relatos de caso com revisão bibliográfica configuram importante fonte de dados para abordagem de experiências diagnósticas e terapêuticas.
No caso, foi apresentado um RN com características morfológicas clássicas da doença. As USG pré-natais sugeriam malformações congênitas e CIUR simétrico, porém o diagnóstico foi definido após o nascimento de acordo com os critérios clínicos recomendados pelo Primeiro Consenso Internacional de Diagnóstico e Manejo da SCdL (2018), sem necessidade de um teste genético. Considerando uma abordagem integral do paciente e sua família, é preciso levar em consideração o real benefício de um teste diagnóstico de alto custo e que não altera prognóstico e condutas posteriores.
O paciente segue em acompanhamento ambulatorial. A puericultura dessas crianças é específica, de acordo com curvas próprias de crescimento e peso. Acompanhar o desenvolvimento psicomotor e a dificuldade de alimentação nesses pacientes é de extrema importância, sendo necessário promover um suporte multidisciplinar cujo objetivo principal é a melhora na qualidade de vida.
REFERÊNCIAS
1. Sarogni P, Pallotta MM, Musio A. Cornelia de Lange syndrome: from molecular diagnosis to therapeutic approach. J Med Genet. 2019 Mai;57(5):289-95.
2. Romano C, Van Wynckel M, Hulst J, Broekaert I, Bronsky J, Dall’Oglio L, et al. European Society for Paediatric Gastroenterology, Hepatology and Nutrition Guidelines for the evaluation and treatment of gastrointestinal and nutritional complications in children with neurological impairment. J Pediatr Gastroenterol Nutr. 2017 Ago;65(2):242-64.
3. Kline AD, Moss JF, Selicorni A, Bisgaard AM, Deardorff MA, Gillett PM, et al. Diagnosis and management of Cornelia de Lange syndrome: first international consensus statement. Nat Rev Genet. 2018 Out;19(10):649-66.
4. Leite AL, Real MV, Santos F. Síndrome de Cornélia de Lange e disgenesia cerebral. Nascer Crescer. 2011;10(4):270-3.
5. Boyle MI, Jespersgaard C, Brondum-Nielsen K, Bisgaard AM, Tumer Z. Cornelia de Lange syndrome. Clin Genet. 2015;88:1-12.
6. Kline AD, Krantz ID, Sommer A, Kliewer M, Jackson LG, Fitzpatrick DR, et al. Cornelia de Lange Syndrome: clinical review, diagnostic and scoring systems, and anticipatory guidance. Am J Med Genet A. 2007 Jun;143A(12):1287-96.
7. De Lange C. Sur un type nouveau de degenerescence (typus Amsterlodamensis). Arch Med Enfants. 1933;36:713-9.
8. Deardorff MA, Noon SE, Krantz ID. Cornelia de Lange syndrome. Seattle: GeneReviews; 2005.
9. Jackson L, Kline AD, Barr MA, Koch S. De Lange syndrome: a clinical review of 310 individuals. Am J Med Genet. 1993;47(7):940-6.
10. Kinjo T, Mekaru K, Nakada M, Nitta H, Hasamoto H, Aoki Y. A case of Cornelia de Lange syndrome: difficulty in prenatal diagnosis. Case Rep Obstetr Gynecol. 2019 Mai;2019:4530491.
11. Dempsey MA, Johnson AEK, Swope BS, Moldenhauer JS, Sroka H, Chong K, et al. Molecular confirmation of nine cases of Cornelia de Lange syndrome diagnosed prenatally. Prenat Diagn. 2014 Fev;34(2):163-7.
12. Urban M, Hartung J. Ultrasonographic and clinical appearance of a 22-week-old fetus with Brachmann‐de Lange syndrome. Am J Med Genet. 2001 Jul;102(1):73-5.
13. Lalatta F, Russo S, Gentilin B, Spaccini L, Boschetto C, Cavalleri F, et al. Prenatal/neonatal pathology in two cases of Cornelia de Lange syndrome harboring novel mutations of NIPBL. Genet Med. 2007 Mar;9:188-94.
14. Bacino CA. Birth defects: approach to evaluation [Internet]. Walthman (MA): UpToDate; 2019; [acesso em 2020 Abr 15]. Disponível em: https://www.uptodate.com/contents/birth-defects-epidemiology-types-and-patterns?search=congenital%20malformations&source=search_result&selectedTitle=1~150&usage_type=default&display_rank=1
15. Kline AD, Barr M, Jackson LG. Growth manifestations in the Brachmann-de Lange syndrome. Am J Med Genet. 1993 Nov;47(7):1042-9.
1. Universidade Federal de Lavras (UFLA), Departamento de Ciências da Saúde (DSA) - Lavras - Minas Gerais - Brasil
2. Santa Casa de Misericórdia de Lavras, UTI Neonatal - Lavras - Minas Gerais - Brasil
Endereço para correspondência:
Ana Elisa Dias de Souza
Universidade Federal de Lavras (UFLA)
Aquenta Sol, Lavras, MG, Brasil. CEP: 37200-900
E-mail: anaelisadias0210@gmail.com
Data de Submissão: 27/04/2020
Data de Aprovação: 06/05/2020
Recebido em: 27/04/2020
Aceito em: 06/05/2020
