Raquitismo é caracterizado por insuficiência ou retardo da mineralização da matriz osteoide, recentemente formada durante o processo de ossificação endocondral, na placa de crescimento. As causas de raquitismo envolvem a deficiência de cálcio, vitamina D e fósforo, bem como as alterações no metabolismo ou na ação da vitamina D1,2.
Existem algumas síndromes que apresentam perda renal isolada de fosfato, resultando em hipofosfatemia, normocalcemia e raquitismo primário3,4. Este quadro clínico é causado por um aumento dos níveis de fator de crescimento fibroblástico-23 derivado do osso (FGF-23) que impede a reabsorção adequada de fosfato no túbulo renal e interfere na hidroxilação renal da vitamina D5,6.
Os genes envolvidos e o modelo de herança desta síndrome podem ser diversos, sendo o raquitismo hipofosfatêmico ligado ao X considerado a causa mais comum de raquitismo hereditário, com uma prevalência variando de 1,7 por 100.00 crianças a 4,8 por 100.000 indivíduos, incluindo adultos e crianças7. Esta forma de apresentação é causada por mutações no gene PHEX, localizado em Xp22.1, que codifica a expressão e degradação do FGF-231,8.
O diagnóstico definitivo da doença é feito através da investigação molecular, porém o exame físico e a avaliação laboratorial e radiológica rotineira podem nortear a investigação diagnóstica, possibilitando o tratamento precoce da doença3.
O tratamento tem como objetivos principais a redução das deformidades esqueléticas e a melhora do ritmo de crescimento, sendo realizado com a reposição de fósforo e calcitriol. A instituição precoce do tratamento é capaz de reduzir a intensidade do retardo de crescimento e das deformidades em membros inferiores3,9.
O burosumab, também indicado nessa condição, é um anticorpo monoclonal com ação em reconhecer e ligar-se à proteína FGF23, bloqueando a sua atividade, o que permite que os rins reabsorvam o fosfato e restaurem os níveis normais de fosfato no sangue. Permite, também, a hidroxilação da vitamina D pelos rins, aumentando os níveis de calcitriol, forma ativa da vitamina D 10,11.
Este trabalho tem como objetivo relatar um caso de uma paciente com raquitismo hipofostâmico ligado ao X, descrevendo as suas características clínicas e laboratoriais da doença, alertando para o diagnóstico precoce desta rara condição.
RELATO DE CASO
Menina, 7 anos e 11 meses, acompanhada pela ortopedia desde 1 ano de idade, devido à deformidade nos membros inferiores. Encaminhada à endocrinologia pediátrica para avaliação.
Na primeira consulta aos 7 anos e 11 meses, apresentava genu varum (Figura 1) e baixa estatura (SDS estatura/idade = -4,8). Exames laboratoriais: fósforo sérico = 2,3mg/dL (4,5-6,6), cálcio iônico = 1,8mmol/L (1,17-1,32), paratormônio = 42pg/mL (12-88), fosfatase alcalina = 600U/L (<300). Iniciada reposição de fósforo e calcitriol, porém sempre apresentou má adesão ao tratamento. A Tabela 1 descreve a evolução clínica e laboratorial da paciente durante o acompanhamento com a endocrinologia pediátrica.
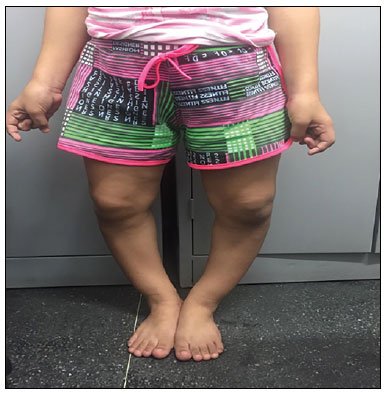 Figura 1. Genu varum em membros inferiores.
Figura 1. Genu varum em membros inferiores.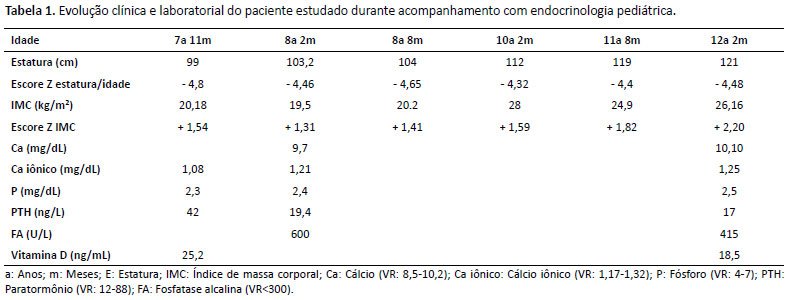
Atualmente, aos 12 anos e 2 meses, permanece com genu varum e baixa estatura (SDS estatura/idade= - 4,48) e queixa de dores ósseas. Exames laboratorias: fosfatase alcalina = 415U/L (<300), cálcio iônico = 1,25mmol/L (1,17-1,32), fósforo sérico = 2,5mg/dL (4-7), paratormônio = 37pg/mL (12-88), vitamina D = 18,5ng/mL (20-280). Foi realizado o sequenciamento do gene PHEX, que evidenciou mutação heterozigótica nesse gene, confirmando o diagnóstico de raquitismo hipofosfatêmico ligado ao X.
Comentários
Os pacientes com raquitismo hipofosfatêmico ligado ao X apresentam altura normal ao nascer e redução da velocidade de crescimento durante os primeiros anos de vida. Após o início da deambulação, as manifestações clínicas típicas da doença se desenvolvem caracterizadas por baixa estatura e deformidades em membros inferiores (genu varum ou genu valgum)1.
Laboratorialmente, a doença manifesta-se por hipofosfatemia, elevação da concentração plasmática de fosfatase alcalina, níveis normais de cálcio sérico, calciúria normal ou diminuída, redução da reabsorção tubular de fósforo e concentração plasmática de paratormônio normal1,9.
As alterações radiológicas são caracterizadas pela perda de definição, alargamento e imagem em cálice observados na zona de calcificação provisória nas metáfises da tíbia, fêmur distal, rádio e ulna1.
A paciente iniciou o acompanhamento em nosso serviço tardiamente, com deformidades ósseas e baixa estatura importantes, além de má adesão ao tratamento. Por estas razões, não houve melhora clínica ou laboratorial nesse período.
REFERÊNCIAS
1. Sociedade Brasileira de Endocrinologia e Metabologia (SBEM). Raquitismo hipofosfatêmico ligado ao X. Projeto Diretrizes. São Paulo: SBEM; 2004.
2. Root AW, Diamond Junior FB. Disorders of calcium metabolism in the child and adolescent. In: Sperling MA, ed. Pediatric endocrinology. 2ª ed. Philadelphia: Saunders; 2002. p. 650-1.
3. Maia MLA, Abreu ALS, Nogueira PCK, Val MLDM, Carvalhaes JTA, Andrade MC. Raquitismo hipofosfatêmico: relato de caso. Rev Paul Pediatr. 2018;36(2):242-7.
4. Mumm S, Huskey M, Cajic A, Wollberg V, Zhang F, Madson KL, et al. PHEX 3’-UTR c.*231A>G near the polyadenylation signal is a relatively common, mild, American mutation that masquerades as sporadic or X-linked recessive hypophosphatemic rickets. J Bone Miner Res. 2015 Jan;30:137-43.
5. White KE, Hum JM, Econs MJ. Hypophosphatemic rickets: revealing novel control points for phosphate homeostasis. Curr Osteoporos Rep. 2014 Sep;12(3):252-62.
6. Goldsweig BK, Carpenter TO. Hypophosphatemic rickets: lessons from disrupted FGF23 control of phosphorus homeostasis. Curr Osteoporos Rep. 2015 Apr;13(2):88-97.
7. Haffner D, Emma F, Eastwood DM, Duplan MB, Bacchetta J, Schnabel D, et al. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat Rev Nephrol. 2019 Jul;15(7):435-55.
8. Ramon-Krauel M. Raquitismo de causa genética. Rev Esp Endocrinol Pediatr. 2018;9(Supl 1):48-53.
9. Gertner JM. Metabolic bone disease. In: Lifshitz F, ed. Pediatric endocrinology. 4a ed. New York: Marcel Dekker; 2003. p. 520;535-6.
10. Carpenter TO, Whyte MP, Imel EA, Boot AM, Högler W, Linglart A, et al. Burosumab therapy in children with X-linked hypophosphatemia. N Engl J Med. 2018 May;378(21):1987-98.
11. Rothenbuhler A, Schnabel D, Högler W, Linglart A. Diagnosis, treatment-monitoring and follow-up of children and adolescents with X-linked hypophosphatemia (XLH). Metabolism. 2020 Feb;103S:153892.
12. Maio P, Mano L, Rocha S, Baptista RB, Francisco T, Sousa H, et al. X-linked hypophosphatemic rickets: a new mutation. J Bras Nefrol. 2021;43(2):279-82.
Hospital Universitário Walter Cantídio, Serviço de Endocrinologia Pediátrica - Fortaleza - Ceará - Brasil
Endereço para correspondência:
Camila Sousa Gonçalves
Hospital Universitário Walter Cantídio
Rua Pastor Samuel Munguba, nº 1290, Rodolfo Teófilo
Fortaleza, CE, Brasil. CEP: 60430-372
E-mail: camilagoncalves_@hotmail.com
Data de Submissão: 16/06/2020
Data de Aprovação: 09/03/2021
Recebido em: 16/06/2020
Aceito em: 09/03/2021
