
A síndrome de Klinefelter (SK) foi descrita em 1940 e é caracterizada por anomalia cromossômica que afeta o sexo masculino, com prevalência estimada de 1 a cada 600 nascidos vivos. Essa desordem cromossômica ocorre pela não disjunção genética na meiose ou mitose durante a gametogênese ou por meio da não disjunção pós-zigótica durante o período embrionário inicial, levando à aneuploidia do cromossomo sexual, apresentando como característica citogenética, em sua grande maioria (90%), um cromossomo X extra (47, XXY); e em sua minoria (10%), moisacismo (46 XY/47XXY), outras aneuploidias raras e mais graves (48 XXXY/ 49 XXXXY) e estruturas cromossômicas anormais1-4. Essa alteração cromossômica pode estar associada ao avanço da idade materna ou paterna5.
Os sinais e sintomas comuns dessa síndrome são consequências da expressão dos genes extras do cromossomo X que podem interromper muitos aspectos do desenvolvimento, como o desenvolvimento sexual antes do nascimento e na puberdade e alterações em estruturas cerebrais6.
Essa síndrome está associada à disfunção testicular cursando frequentemente com hipogonadismo e/ou infertilidade por consequência de uma falência androgênica. Os achados clínicos principais são volume testicular reduzido (máximo de 10 ml), azoospermia e aumento das gonadotrofinas. Pode cursar também com ginecomastia, atraso puberal, pilificação diminuída, micropênis, alta estatura, aumento da envergadura em relação à estatura, dificuldade de aprendizagem e fala, obesidade, doenças psiquiátricas associadas à imagem, síndrome metabólica, doenças autoimunes e câncer1-3.
Na presença de hipogonadismo associado a baixos níveis de testosterona, a terapia de reposição hormonal continua sendo o tratamento médico de escolha e sua dose é individualizada. Esse tratamento não traz mudanças dos padrões corporais já estabelecidos e déficits de desenvolvimento neurológico, mas apresenta melhora no bem-estar, atenção e humor4,5.
Apesar de bem estabelecidas as características fenotípicas da síndrome, é estimado que apenas 25% a 35% dos indivíduos do sexo masculino portadores de SK são diagnosticados ao longo da vida devido à variabilidade na apresentação clínica. Visto o baixo percentual de diagnóstico na infância e adolescência, o trabalho objetiva alertar a importância da investigação das alterações testiculares associadas ou não a outros sinais de hipogonadismo para diagnóstico e tratamento antes da fase adulta.
O estudo realizado é de caráter descritivo, realizado em unidade de atendimento infantil em um município no interior de São Paulo; o estudo respeita os princípios da resolução nº 510/2016, da Comissão Nacional de Saúde e foi aprovado pela comissão de ética e pesquisa da Universidade de Taubaté sob o número CAAE: 37218120.0.0000.5501.
RELATO DE CASO
JPAL, 16 anos, masculino, solteiro, estudante, acompanhado pela tia, foi encaminhado à consulta de Hebiatria para acompanhamento e orientações sobre sexualidade.
Paciente referia ginecomastia unilateral à esquerda há 2 anos e testículos pequenos, essa última informação não havia sido relatada à responsável. Adolescente cuidado pela tia, possuía história pregressa de pneumonia aos 7 anos e litíase renal aos 14 anos, sem alergias ou cirurgias prévias. Jovem já em programação de correção cirúrgica de ginecomastia.
Indivíduo tímido, porém com boa interação social, comportamento alimentar e hábitos de vida compatíveis com período da adolescência, negava uso de medicações e/ou drogas lícitas e ilícitas, sem histórico familiar de doenças genéticas e/ou cardiovasculares.
Ao exame físico, apresentava biotipo eunucoide (longilíneo com predomínio do segmento inferior), com estatura de 177 cm (z- score entre 0 e +1), envergadura de 180,5 cm, peso de 67,5 kg, IMC 21,54 kg/m2 (z- score entre 0 e +1). O aparelho genitourinário com distribuição dos pêlos em padrão masculino, testículos impúberes, critério para maturação sexual Marshall & Tanner G1 P4, com evidência de ginecomastia grau dois unilateral à esquerda. O exame dos demais aparelhos não apresentava alterações, exceto pela pressão arterial aferida no momento da consulta de 136 x 90 mmHg, compatível com hipertensão estágio 1.
Realizada ultrassonografia de bolsa testicular que apresentava testículos com dimensões reduzidas, ambos com volume de 1,7 cm3, texturas homogêneas, epidídimos de forma, ecogenicidade e espessuras normais, sem evidências de lesões focais sólidas e/ou císticas em testículos e epidídimos. Cordões espermáticos de aspecto normal (figura 1).

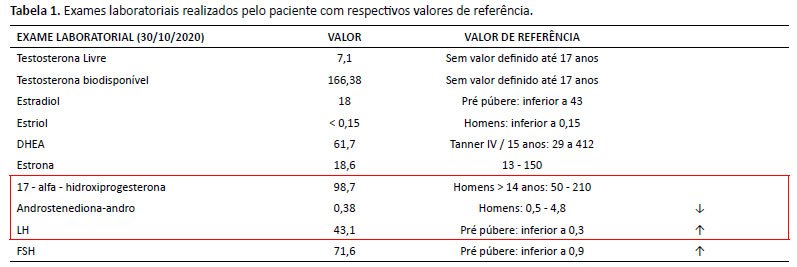
Os exames laboratoriais revelaram aumento de hormônio folículo estimulante (FSH) e hormônio luteinizante (LH), e redução de androstenediona (Tabela 1). O exame do cariótipo banda G revelou 47, XXY, confirmando a hipótese diagnóstica de Síndrome de Klinefelter.
Realizadas orientações ao adolescente em relação à sexualidade e possíveis repercussões pela síndrome apresentada, bem como encaminhamento à psicologia e endocrinologia pediátrica com novos pedidos de exames laboratoriais para avaliar a necessidade de reposição hormonal.
DISCUSSÃO DO CASO
A SK é uma desordem genética comum que cursa com insuficiência endócrina, apresentando variações nas concentrações de testosterona sérica e variabilidade fenotípica, o que faz com que seja subdiagnosticada.
Possíveis características clínicas em adolescentes com SK descritas na literatura são: testículos pequenos e sólidos, virilização diminuída, ginecomastia, gordura abdominal, disfunção sexual, problemas de imagem corporal e humor deprimido. Além desses sinais e sintomas, podem ser encontrados início tardio da puberdade, alta estatura, aumento teor de gordura5. No caso em questão, o adolescente apresentava ginecomastia, testículos pequenos e firmes, alta estatura, hipertensão e distúrbio de imagem corporal, compatíveis com a literatura.
Em artigo publicado por Hovnik et al. (2022)7, foi relatado o caso de um paciente que foi referenciado ao serviço devido à ginecomastia e, durante avaliação clínica, foram evidenciados constituição astênica, testículos pequenos e tópicos, história prévia de correção cirúrgica de hipospádia e orquipexia. Investigação laboratorial demonstrava hipogonadismo associado à queda de testosterona e cariótipo 47 XXY/ 46XX, sendo confirmado mosaicismo para SK. No presente relato de caso, o motivo do encaminhamento foi semelhante e os achados do exame físico nortearam o diagnóstico, o qual foi corroborado através do cariótipo.
Importante salientar que a ginecomastia encontrada no caso reportado apresentava evolução maior que 18 meses, sem sinais de regressão espontânea, associada à alteração em gônadas, o que descaracteriza a ginecomastia fisiológica.
A vivência da adolescência é permeada pela importante mudança na composição corporal, evolução da maturidade e identidade sexual, o que pode trazer consequências psicossociais se tais modificações não estiverem dentro da expectativa do adolescente, o que ocorreu com o paciente deste caso. Como descrito na literatura, o acompanhamento psicológico é útil para melhorar essas experiências e oferecer oportunidades para desenvolver estratégias para reconhecer, processar e expressar sentimentos e pensamentos. Meninos com SK correm maior risco de comprometimento da cognição social e percepções menos precisas de pistas socioemocionais. O conceito de prováveis distúrbios de fertilidade deve ser debatido juntamente com revisões regulares da puberdade e função sexual em adolescentes8, como foi feito neste caso. A importância do diagnóstico correto e precoce é, justamente, poder atuar na prevenção e/ou tratamento de suas consequências imediatamente, como micropênis e testículos pequenos, dificuldades de aprendizagem, puberdade tardia, infertilidade, alta estatura, esqueleto eunucoide, ginecomastia, osteoporose e outros distúrbios metabólicos, melhorando a qualidade de vida do indivíduo.
CONCLUSÃO
Neste trabalho apresentamos um caso de SK diagnosticado durante a puberdade. Apesar de ser uma desordem genética comum, ainda apresenta poucos diagnósticos antes da idade adulta e, para tanto, precisa ser identificada por pediatras e hebiatras para determinação precoce, preferencialmente na fase pré-puberal, proporcionando acompanhamento multidisciplinar e intervenções, quando necessárias, já na infância e puberdade. O reconhecimento pode ser feito por meio de triagem simples através da palpação e estimativa de volume testicular nas consultas de rotina de todos os meninos impúberes e púberes.
REFERÊNCIAS
1. Tincani BJ, Mascagni BR, Pinto RDP, Guaragna-Filho G, Castro CCTS, Sewaybricker LE, et al. Klinefelter syndrome: an unusual diagnosis in pediatric patients. J Pediatr (Rio J). 2012;88(4):323-7.
2. Bearelly P, Oates R. Recent advances in managing and understanding Klinefelter syndrome. F1000Res. 2019;8(F1000 Faculty Rev):112.
3. Davis S, Howell S, Wilson R, Tanda T, Ross J, Zeitler P, et al. Advances in the Interdisciplinary Care of ChildreN with Klinefelter Syndrome. Adv Pediatr. 2016;63(1):15-46.
4. Lizarazo AH, McLoughlin M, Vogiatzi MG. Endocrine aspects of Klinefelter syndrome. Curr Opin Endocrinol Diabetes Obes. 2019;26(1):60-5.
5. Zitzmann M, Aksglaede L, Corona G, Isidori AM, Juul A, T'Sjoen G, et al. European Academy of Andrology Guidelines on Klinefelter Syndrome Endorsing Organization: European Society of Endocrinology. Andrology. 2012;9(1):145-67.
6. Curado RMOF, Sestari SJ, Gamba BF, Bicudo LAR, Approbato MS, Amaral WN, et al. Síndrome de Klinefelter, uma condição subdiagnosticada: revisão de literatura. RRS FESGO. 2020 jan.-jul.;3(1):68-75
7. Hovnik T, Zitnik E, Avbelj Stefanija M, Bertok S, Sedej K, Bancic Silva V, et al. An Adolescent Boy with Klinefelter Syndrome and 47,XXY/46,XX Mosaicism: Case Report and Review of Literature. Genes (Basel). 2022 Apr 23;13(5):744. DOI: https://doi.org.10.3390/genes13050744.
8. Butler G, Srirangalingam U, Faithfull J, Sangster P, Senniappan S, Mitchell R. Klinefelter syndrome: going beyond the diagnosis. Arch Dis Child. 2023 Mar;108(3):166-71.
Universidade de Taubaté, Residência Médica - Pediatria - Taubaté - São Paulo - Brasil
Endereço para correspondência:
Yuki Horigome
Universidade de Taubaté
Rua Quatro de Março, 432, Reitoria, Centro
Taubaté, SP, Brasil. CEP: 12020-270
E-mail: yuki_horigome@hotmail.com
Data de Submissão: 20/09/2023
Data de Aprovação: 19/03/2025
