A síndrome CDAGS (craniossinostose, surdez, anomalias anais, geniturinárias e cutâneas) é uma condição rara, autossômica recessiva, com poucos relatos na literatura, caracterizada por craniossinostose, fechamento retardado das fontanelas, perda auditiva, anomalias anais, malformações geniturinárias e poroceratose1. Estudos recentes vêm mostrando associação da síndrome com áreas cromossômicas específicas. No entanto, até o momento, os genes envolvidos ainda não foram identificados2.
As lesões cutâneas são descritas como placas eritematodescamativas reticuladas, com bordas definidas e crostas na superfície, sendo semelhante, tanto clinicamente quanto histologicamente, à poroceratose. Deve-se ter atenção na presença desse quadro cutâneo, pois ele está frequentemente presente na síndrome CDAGS, devendo essa hipótese diagnóstica ser aventada e outras alterações clínicas da síndrome investigadas. Avaliação genética também deve ser considerada na abordagem diagnóstica3.
O objetivo deste estudo foi descrever os achados clínicos, alertar sobre a existência dessa enfermidade e sobre a necessidade em suscitar a síndrome CDAGS entre os diagnósticos diferenciais na presença de acometimento cutâneo semelhante à poroceratose, associado a outras alterações características da síndrome. O reconhecimento da doença e de suas alterações clínicas possibilita um diagnóstico oportuno, evitando prescrição de tratamentos ineficazes com efeitos colaterais e permitindo acompanhamento multidisciplinar e tratamento adequado mais precoces, melhorando a condição e a qualidade de vida dos pacientes.
RELATO DE CASO
Recém-nascido (RN), a termo, sexo masculino, nascido de parto cesárea em dezembro de 2022. Em seu primeiro exame físico, observaram-se fácies típicas (fenda palpebral oblíqua, hipertelorismo, raiz nasal alta e ampla, orelhas rodadas posteriormente com sobredobramento de hélix, boca em carpa, palato alto e ogival e retrognatia), craniossinostose associada a turribraquicefalia, clinodactilia, hipospadia, escroto em cachecol bífido e ânus imperfurado. Além disso, apresentou o exame de emissões otoacústicas evocadas alterado bilateralmente, sendo confirmada a deficiência auditiva pelo exame do potencial evocado auditivo do tronco encefálico (BERA).
Do nascimento até os 7 meses de vida, o paciente foi submetido a inúmeras cirurgias para correção da imperfuração anal associada à fístula anorretal, pois apresentou recorrência de deiscência de sutura como complicação pós-operatória. Além disso, foi submetido ao tratamento cirúrgico para correção da craniossinostose aos 5 meses de vida. Na análise do cariótipo não se identificou anormalidade cromossômica, sendo o resultado 46,XY. Então, complementou-se a análise genética com a técnica de hibridização genômica comparativa por microarray, a qual não detectou microdeleções ou microduplicações no material genético.
Aos 3 meses de vida, iniciou com placas eritematodescamativas pruriginosas de figuração circinada, confluentes, nos membros, dorso de mãos e pés, poupando palmas, plantas e dobras. Mãe notava leve melhora das lesões com exposição solar. Para as lesões de pele, foram prescritos diferentes corticoides e antibióticos tópicos, além de ácido salicílico a 3% pomada, sem aparente melhora no aspecto das lesões. Além disso, fez uso de diversos antibióticos sistêmicos, tanto via oral quanto parenteral, ficando em duas oportunidades internado somente para recebimento endovenoso da medicação. Ainda, foi prescrito hidroxizina para alívio do prurido e de zinco quelado, por período determinado de tempo, por suspeita de acrodermatite enteropática.
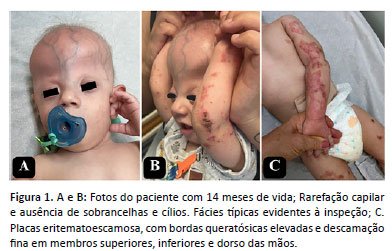
Aos 14 meses, foi encaminhado para primeira consulta com a Dermatologia. Percebeu-se xerose cutânea leve, associada a placas numulares escamosas e eritematosas com bordas queratósicas elevadas e descamação fina em membros, dorso de mãos e pés. Ainda, notou-se rarefação capilar e ausência de sobrancelhas e de cílios (figura 1). Recebeu orientação de uso de creme hidratante em todo corpo de 2 a 3 vezes por dia e realizou biópsia incisional das lesões de pele. O anatomopatológico revelou um padrão histológico muito semelhante à poroceratose, com lesão de interface, queratinócitos apoptóticos, áreas de disqueratose, hipergranulose, hiperqueratose (figura 2). Após o resultado da biópsia de pele e após pesquisa na literatura, a hipótese da síndrome CDAGS foi aventada.

A síndrome CDAGS é uma doença rara, autossômica recessiva, cujo acrônimo foi proposto por Mendoza-Londono et al. (2005)1. A sigla resume os principais sinais e sintomas da síndrome: craniossinostose e hipoplasia clavicular (letra C); fechamento retardado da fontanela e, em alguns pacientes, surdez (letra D); anomalias anais, como atresia anal e ânus ectópico anterior (letra A); malformação geniturinária, como hipospadia e fístula uretro-retal (letra G) e erupção cutânea, a qual, em alguns casos, pode ser classificada como poroceratose (letra S)1,4.
Em função da raridade da doença, há relatos de apenas treze casos de diferentes regiões geográficas de 1981 até 2017. Embora o padrão de herança seja consistente com uma doença autossômica recessiva, os genes responsáveis ainda não foram identificados com exatidão5. Acredita-se que a síndrome CDAGS seja uma doença autossômica recessiva. Estudos recentes vêm mostrando a possível associação com áreas cromossômicas. No entanto, até o momento, os genes envolvidos nessa síndrome ainda não foram identificados3.
O quadro cutâneo está frequentemente presente na síndrome CDAGS. Com isso, na presença de lesões de pele semelhantes à poroceratose, outras alterações clínicas como craniossinostose, fechamento retardado das fontanelas, malformações geniturinárias e anomalias anais devem ser prontamente investigadas para um diagnóstico precoce. O atraso diagnóstico acarreta angústia e sofrimento para as famílias e está associado a altas taxas de morbidade evitável. O diagnóstico precoce possibilita melhores escolhas terapêuticas, como abordagem cirúrgica oportuna das malformações, reduzindo as complicações relacionadas ao quadro tardio e provendo um melhor prognóstico para os portadores dessa síndrome. No caso do paciente do relato, por exemplo, o manejo cirúrgico precoce da craniossinostose e da imperfuração anal evitou complicações como hipertensão intracraniana e formação de fístulas, respectivamente.
As lesões cutâneas são principalmente descritas como placas e pápulas eritematoescamosas, pleomórficas, com bordas nítidas, elevadas e ceratóticas, semelhantes à poroceratose, podendo conter crostas hemáticas, como no nosso paciente2,4. Alopecia do couro cabeludo e escassez ou ausência de sobrancelhas e cílios também são descritos na síndrome de CDAGS, alterações as quais nosso paciente do relato também apresenta2.
Histologicamente, a síndrome CDAGS também é semelhante à poroqueratose, com degeneração vacuolar focal da camada basal e áreas de disqueratose3. Ainda não está claro se as lesões cutâneas na síndrome CDAGS representam poroceratose clássica ou se são apenas notoriamente semelhantes. Consideramos que tais lesões seriam melhor classificadas dentro do atual grupo das doenças autoinflamatórias queratinizantes no qual se inserem, por exemplo, a própria poroqueratose e a queratose liquenoide crônica, que têm em comum um padrão de resposta tecidual liquenoide, associado a distúrbios da queratinização. Embora sejam histologicamente semelhantes, podem comportar-se de formas diferentes, com as lesões cutâneas da síndrome de CDAGS apresentando melhora com a exposição solar, enquanto as da poroceratose sabidamente pioram com a exposição à luz ultravioleta6,7.
A raridade da síndrome implica atraso diagnóstico e consequentes tentativas de abordagens terapêuticas ineficazes. Alterações como ânus imperfurado ou fístula anorretal podem ser tratadas cirurgicamente e, geralmente, são abordadas precocemente4. Na craniossinostose coronal, o tratamento cirúrgico é indicado, no caso de sinostose coronal, entre 6-9 meses para correção da deformidade do crânio e pelo risco de hipertensão intracraniana. Procedimentos adicionais, como injeção de gordura no esqueleto craniofacial para diminuir assimetrias faciais e correção de ptose palpebral, podem ser necessários2,3,8. Já no caso do quadro cutâneo, diversos tratamentos são prescritos na falta do diagnóstico correto, como esteroides locais e sistêmicos, antibióticos, antifúngicos, retinoides sistêmicos e suplementação de zinco e biotina, sem sucesso. O manejo não farmacológico, com hidratação diária da pele, é importante na integridade da barreira cutânea, evitando infecções secundárias e prevenindo a penetração de alérgenos e outros agentes irritantes2. Além disso, o manejo das lesões cutâneas ainda deve incluir o tratamento de infecções secundárias, quando presentes, e a vigilância da pele devido ao risco potencial de carcinoma de células escamosas, como na poroceratose7.
Devido ao pequeno número de casos notificados até o momento, a síndrome CDAGS continua a ser um desafio diagnóstico. Como parte da abordagem diagnóstica, a avaliação genética deve ser considerada. Embora alguns trabalhos tenham mostrado a possível associação de uma região cromossômica com a doença, ainda não foram identificados com precisão os genes causadores da síndrome. Por isso, são necessários mais estudos para estabelecer a sua etiologia. Por fim, com objetivo de ampliar o conhecimento e reforçar a existência da enfermidade para a comunidade médica, relatamos o caso de um paciente com as principais alterações descritas na sigla da síndrome, chamando atenção para as manifestações cutâneas, as quais quase sempre estão presentes no quadro. Ainda que, na síndrome CDAGS, as malformações possam sofrer correção cirúrgica, como nas anomalias anais, as demais alterações clínicas não abordáveis cirurgicamente, incluindo as manifestações cutâneas, impactam negativamente na qualidade de vida dos pacientes. Portanto, a detecção oportuna da síndrome permitirá o início precoce do tratamento que poderá melhorar a condição de vida e o bem-estar dos pacientes.
REFERÊNCIAS
1. Mendoza-Londono R, Lammer E, Watson R, Harper J, Hatamochi A, Hatamochi-Hayashi S, et al. Characterization of a new syndrome that associates craniosynostosis, delayed fontanel closure, parietal foramina, imperforate anus, and skin eruption: CDAGS. Am J Hum Genet. 2005;77(1):161-8. DOI: https://doi.org/10.1086/431654.
2. Pastrana-Ayala R, Peña-Castro GL, Valencia-Herrera AM, Mena-Cedillos CA, Toussaint-Caire S, Akaki-Carrño YI, et al. Craniosynostosis, delayed closure of the fontanelle, anal, genitourinary, and skin abnormalities (CDAGS syndrome): first report in a Mexican patient and review of the literature. Int J Dermatol. 2017;56(4):435-9. DOI: https://doi.org/10.1111/ijd.13504.
3. Xing C, Kanchwala M, Rios JJ, Hyatt T, Wang RC, Tran A, et al. Biallelic variants in RNU12 cause CDAGS syndrome. Hum Mutat. 2021;42(8):1042-52. DOI: https://doi.org/10.1002/humu.24239.
4. Chouery E, Guissart C, Mégarbané H, Aral B, Nassif C, Thauvin-Robinet C, et al. Craniosynostosis, anal anomalies, and porokeratosis (CDAGS syndrome): case report and literature review. Eur J Med Genet. 2013;56(12):674-7. DOI: https://doi.org/10.1016/j.ejmg.2013.09.012.
5. Cohen-Sors R, Devauchelle B, Vabres P, Jain M, Demeer B, Carmi E. Syndrome CDAGS (craniosténose, surdité, anomalie anale et génito-urinaire avec éruption cutanée) [CDAGS syndrome (craniostenosis, deafness, anal abnormalities and genitourinary malformations with skin rash)]. Ann Dermatol Venereol. 2020;147(12):868-72. DOI: https://doi.org/10.1016/j.annder.2020.10.016.
6. Blicharz L, Czuwara J, Rudnicka L, Torrelo A. Autoinflammatory Keratinization Diseases-The Concept, Pathophysiology, and Clinical Implications. Clin Rev Allergy Immunol. 2023;65(3):377-402. DOI: https://doi.org/10.1007/s12016-023-08971-3.
7. Taylor A, Nguyen RH, Glass DA 2nd, Agim NG. JAAD Grand Rounds. Annular plaques and craniosynostosis. J Am Acad Dermatol. 2013;68(5):881-4. DOI: https://doi.org/10.1016/j.jaad.2011.12.005.
8. Ghizoni E, Denadai R, Raposo-Amaral CA, Joaquim AF, Tedeschi H, Raposo-Amaral CE. Diagnosis of infant synostotic and nonsynostotic cranial deformities: a review for pediatricians. Rev paul pediatr. 2016;34(4):495-502. DOI: https://doi.org/10.1016/j.rppede.2016.02.005.
1. Hospital Universitário da Universidade Federal de Santa Catarina (HU-UFSC), Serviço de Dermatologia - Florianópolis - Santa Catarina - Brasil
2. Hospital Universitário da Universidade Federal de Santa Catarina (HU-UFSC), Serviço de Patologia - Florianópolis - Santa Catarina - Brasil
3. Hospital Infantil Joana de Gusmão (HIJG), Serviço de Dermatologia Pediátrica - Florianópolis - Santa Catarina - Brasil
Endereço para correspondência:
Isabel Crivelatti
Hospital Universitário da Universidade Federal de Santa Catarina (HU-UFSC).
Rua Profa. Maria Flora Pausewang, Trindade,
Florianópolis, SC, Brasil. CEP: 88036-800.
E-mail: isabel.crivelatti@gmail.com
Data de Submissão: 28/10/2024
Data de Aprovação: 05/04/2025
Recebido em: 28/10/2024
Aceito em: 05/04/2025
