Linfohistiocitose hemofagocítica (HLH) é uma doença caracterizada pela desregulação do sistema imunológico em consequência da disfunção dos linfócitos T CD8+ e das células natural-killer (NK)1. Essa condição favorece para a produção excessiva de citocinas inflamatórias, podendo resultar em óbito se não tratada de forma precoce. Pode ser classificada em primária (familiar ou doença genética) ou secundária (adquirida)1,2. Entre as causas secundárias, o vírus Epstein-Barr (EBV) é o agente infeccioso mais associado com a doença3,4.
Atualmente, 5 de 8 critérios são necessários para fazer o diagnóstico: febre; citopenias (pelo menos duas linhagens); esplenomegalia; hiperferritinemia; hipertrigliceridemia e/ou hipofibrinogenemia; hemofagocitose em medula óssea, linfonodo ou baço; níveis de CD-25 elevados; e redução/ausência da atividade de células NK1. Entretanto, estudos têm apontado que alguns pacientes podem não apresentar todos os critérios necessários para diagnóstico no início do quadro, com aparecimento dos mesmos em tempos diferentes durante o curso da doença, o que pode impactar no início tardio da terapia4. Assim, deve ser feito alto nível de suspeição para aumentar as chances de sobrevida desses pacientes (Tabela 1).

O presente trabalho tem como objetivo apresentar o caso de uma criança com HLH associado com EBV, de curso clínico grave, que resultou em desafio diagnóstico para a equipe assistente. O trabalho foi aprovado por um comitê de ética em pesquisa.
RELATO DE CASO
Paciente do sexo masculino, 5 anos, previamente hígido, com cartão vacinal completo, apresentou quadro de febre diária, 2 picos ao dia, associado com odinofagia por 10 dias antes da internação. Nega tosse ou contactantes doentes. Durante período, foi avaliado por 3 pediatras, sendo prescrito antibioticoterapia, sem melhora. Evoluiu com aumento e dificuldade de mobilização da região cervical, o que motivou os pais a procurarem a unidade de emergência.
Na admissão, foram identificados sinais de sepse, aumento da região cervical, com presença de nódulos duros e dolorosos à palpação local, e hepatoesplenomegalia. Não foram observadas alterações neurológicas, oftalmológicas, orofaríngeas, dermatológicas ou osteoarticulares. Os exames laboratoriais evidenciaram hemoglobina: 12,4g/dL, leucócitos: 364/mm3, plaquetas: 490,000/mm3, proteína C-reativa: 325mg/dL, aspartato aminotranferase (AST): 93U/L, alanina aminotranferase (ALT): 60U/L e albumina: 2,4g/dL. Coagulograma, bilirrubinas, creatinina e radiografia de tórax sem alterações. Optou-se por iniciar tratamento para neutropenia febril e solicitar vaga de internação em unidade de terapia intensiva (UTI).
No 3º dia após a admissão, o paciente evoluiu com quadro de choque hemodinâmico com disfunção de múltiplos órgãos, apresentando AST: 9.043U/L, ALT: 2.584U/L, bilirrubina direta: 2,58g/dL, bilirrubina indireta: 0,36g/dL, lactato desidrogenase: 37.321U/L, creatinina: 1mg/dL, plaquetas: 136mil/mm3, tempo de protrombina (TP) incoagulável, tempo de tromboplastina parcial ativada (TTPa) incoagulável e baixos níveis dos complementos C3 e C4. O paciente foi tratado com antibiótico de amplo espectro, drogas vasoativas, terapia de substituição renal, ventilação mecânica e terapia transfusional.
Para investigação, foram coletados anticorpos (IgM e IgG) para vírus Epstein-Barr (EBV), citomegalovírus, herpes simples, vírus da imunodeficiência humana, hepatite B, hepatite C, toxoplasmose, bartonella hanseleae e sífilis, sendo todos não reagentes. Além disso, foi realizada investigação para doenças reumatológicas (fator reumatoide, fator antinuclear, anti-DNA dupla-hélice, anti-Smith, anti-coagulante lúpico, anti-cardiolipina, anti-citoplasma de neutrófilos c e p, anti-Ro, anti-La e Coombs), apresentando resultados normais. Não foi identificado alterações das imunoglobulinas séricas. Houve crescimento de Pseudomonas aeruginosa na hemocultura de sangue periférico durante a internação.
Com relação aos exames de imagem, a tomografia de região cervical confirmou presença de linfonodos aumentados bilateralmente, sem evidência de abcesso ou outras alterações. A tomografia de tórax e o ecocardiograma apresentaram resultados normais. A tomografia de abdome evidenciou hepatoesplenomegalia e ascite discreta. A histopatologia e imunofenotipagem de linfonodo cervical (duas amostras em tempos diferentes) mostraram proliferação linfoide atípica com necrose importante, não sendo conclusivo para linfoma. O aspirado de medula óssea apresentou agranulocitose, sem outras alterações.
Diante do apresentado, a principal hipótese foi linfohistiocitose hemofagocítica, sendo reforçada pela presença de hipertrigliceridemia (466mg/dL) e hiperferritinemia (1286μg/L). Posteriormente, foi confirmada infecção associada por vírus Epstein-Barr através de reação de polimerase em cadeia (PCR) em sangue periférico. A terapêutica específica com corticoterapia associada com ciclosporina foi iniciada no 7º dia de internação, apresentando melhora clínica-hemodinâmica. Entretanto, na terceira semana de tratamento, o paciente cursou com choque hipotensivo refratário associado com coagulação intravascular disseminada, evoluindo com hemorragia pulmonar e óbito no 28º dia de internação hospitalar.
COMENTÁRIOS
A HLH é uma doença grave, devendo o diagnóstico ser realizado de forma precoce. A abordagem de investigação envolve diagnóstico diferencial de adenomegalia cervical e febre. A associação com disfunção hepática e citopenias, após excluir causas infecciosas, oncológicas e reumatológicas, aponta para essa doença5 (Tabela 2).
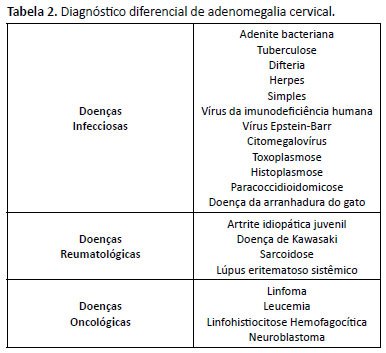
Com relação aos critérios diagnósticos de HLH, nosso paciente apresentava febre, esplenomegalia e neutropenia no início do quadro, sendo identificado plaquetopenia, hipertrigliceridemia e hiperferritinemia durante a internação, atrasando a terapia em 7 dias. Há relatos de até 67 dias de internação até a confirmação diagnóstica6.
Xu et al. (2017)7, após analisar retrospectivamente 323 pacientes com HLH, identificaram presença de febre (100%), hiperferritinemia (92,8%), esplenomegalia (73,7%), plaquetopenia (70,7%), hemofagocitose (68,1%), hipofibrinogenemia (57,9%) e neutropenia (56,8%) entre os critérios mais frequentes. Outros trabalhos apresentaram resultados semelhantes6,8.
Em recente publicação da Sociedade Europeia de Infectologia Pediátrica, foi abordado sobre a dificuldade de confirmação de HLH em alguns pacientes, pela ausência de critérios diagnósticos no início do quadro, com aparecimento durante o curso da doença9. Associado a isso, existe baixa disponibilidade de alguns exames, como contagem de CD-25, atividade de células NK e sequenciamento genético, mesmo em se tratando de grandes centros7,9.
O paciente do caso desenvolveu HLH secundária à infecção por EBV, sendo o agente infeccioso mais associado com a doença3,4,9. A confirmação da infecção ocorreu apenas através de PCR viral. Imashuku et al. (2002)10 relataram que o melhor exame para diagnóstico de EBV, em pacientes com HLH, é o PCR viral, já que 61% dos exames sorológicos apresentam resultados falso-negativos.
Outras manifestações atípicas do EBV podem ser encontradas nesse caso, o que resultou em dificuldade na interpretação dos achados. Agranulocitose em medula óssea já foi relatada em outros pacientes com esse tipo de infecção, justificando o quadro inicial de neutropenia grave3,11. A hemofagocitose, marca característica da HLH, pode não ocorrer na fase inicial da doença, não sendo sensível para realização do diagnóstico6. Além disso, pacientes portadores de HLH por EBV podem apresentar proliferação linfoide atípica com necrose na biópsia de linfonodo cervical10.
Atualmente, não existem fortes evidências sobre o melhor tratamento para HLH por EBV, sendo os protocolos baseados em consensos de especialistas, devendo cada caso ser avaliado de forma individualizada. No tratamento inicial, pode ser utilizado imunoglobulina intravenosa ou corticoterapia associado com ciclosporina, reservando o uso do etoposídeo para casos mais graves, segundo protocolo HLH-20041,2; outra opção é o uso do rituximab9. Devido ao quadro de choque descompensado com uso de drogas vasoativas em altas doses, optou-se por não fazer uso do etoposídeo, no caso descrito, pelo risco de deterioração clínica.
CONCLUSÃO
É necessário alto nível de suspeição para realizar o diagnóstico de linfohistiocitose hemofagocítica de forma precoce, apesar da raridade da doença, para possibilitar maior sobrevida aos pacientes que venham a desenvolver esse problema.
REFERÊNCIAS
1. Henter JI, Horne AC, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007 Fev;48(2):124-31.
2. Ehl S, Astigarraga I, Greenwood TVB, Hines M, Home AC, Ishii E, et al. Recommendations for the use of etoposide-based therapy and bone marrow transplantation for the treatment of HLH: consensus statements by the hlh steering committee of the histiocyte society. J Allergy Clin Immunol Pract. 2018 Set/Out;6(5):1508-17.
3. Brito EES, Moreira LAC. Síndrome de mononucleose infecciosa com sorologia positiva para citomegalovírus e Epstein-Barr vírus. Resid Pediatr. 2016;6(1):31-4.
4. Domachowske JB. Infectious triggers of hemophagocytic syndrome in children. Pediatr Infect Dis J. 2016 Nov;25(11):1067-8.
5. Matos LL, Faro Junior MP, Kanda JL, Gerardi Filho VA, Fernandes PM. Linfadenopatia cervical na infância: etiologia, diagnóstico diferencial e terapêutica. Arq Bras Ciên Saúde. 2010 Set/Dez;35(3):213-9.
6. Oguz MM, Sahin G, Acoglu EA, Polat E, Yucel H, Celebi FZO, et al. Secondary hemophagocytic lymphohistiocytosis in pediatric patients: a single center experience and factors that influenced patient prognosis. Pediatr Hematol Oncol. 2019 Fev;36(1):1-16.
7. Xu XJ, Wang HS, Ju XL, Ziao PF, Xiao Y, Xue HM, et al. Clinical presentation and outcome of pediatric patients with hemophagocytic lymphohistiocytosis in China: a retrospective multicenter study. Pediatr Blood Cancer. 2017 Abr;64(4):e26264.
8. Karapinar B, Yilmaz D, Balkan C, Akin M, Ay Y, Kvakli K. An unusual cause of multiple organ dysfunction syndrome in the pediatric intensive care unit: hemophagocytic lymphohistiocytosis. Pediatr Crit Care Med. 2009 Mai;10(3):285-90.
9. Chesshyre E, Ramanan A, Rodrick MR. Hemophagocytic lymphohistiocytosis and infections: an update. Pediatr Infect Dis J. 2019 Mar;38(3):e54-e6.
10. Imashuku S. Clinical features and treatment strategies of Epstein-Barr virus associated hemophagocytic lymphohistiocytosis. Crit Rev Oncol Hematol. 2002 Dez;44(3):259-72.
11. Bolis V, Karadedos C, Chiotis I, Chaliasos N, Tsabouri S. Atypical manifestations of Epstein-Barr virus in children: a diagnostic challenge. J Pediatr (Rio J). 2016 Mar/Abr;92(2):113-21.
Hospital Pequeno Príncipe, Unidade de Terapia Intensiva Pediátrica - Curitiba - Paraná - Brasil
Endereço para correspondência:
Jáder Pereira Almeida
Hospital Pequeno Príncipe
Rua Desembargador Motta, nº 1070, Água Verde
Curitiba - PR. Brasil. CEP: 80250-060
E-mail: jader.pa@hotmail.com
Data de Submissão: 29/07/2019
Data de Aprovação: 10/09/2019
Recebido em: 29/07/2019
Aceito em: 10/09/2019
